
User Experience#
One of the main barriers towards using the molecular and materials simulation codes is its demanding learning curve and required expertise in working with the interfaces. The main reason behind the aforementioned problem stems from the intricate nature of the software engines and how to control their performance on processing the input data. What makes this situation worse in the lack of global standardizations and software-specific syntaxes and keywords for setting up various simulation parameters.
The following script is an input file for the GROMACS program package which sets up the necessary parameters for a 1 ns constant temperature-constant pressure (NPT) molecular dynamics simulation:
integrator = md
dt = 0.002
nsteps = 500000
nstlog = 5000
nstenergy = 5000
nstxout-compressed = 5000
continuation = yes
constraints = all-bonds
constraint-algorithm = lincs
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = Cut-off
rvdw = 1.0
DispCorr = EnerPres
tcoupl = V-rescale
tc-grps = Protein SOL
tau-t = 0.1 0.1
ref-t = 300 300
pcoupl = Parrinello-Rahman
tau-p = 2.0
compressibility = 4.5e-5
ref-p = 1.0
A similar input file for LAMMPS will look like the following:
units real
boundary p p p
atom_style full
newton on
pair_style lj/class2/coul/long 10.0
pair_modify mix sixthpower tail yes shift no
kspace_style pppm 1e-05
bond_style hybrid class2 harmonic
angle_style hybrid class2 harmonic
dihedral_style hybrid class2 harmonic
improper_style class2
read_data structure.dat
timestep 2.0
fix 1 all temp/rescale 500 300.0 300.0 20.0 1.0
fix 2 all nve
fix 3 all ave/time 25 1 25 v_time v_temp v_press v_density v_cella \
v_cellb v_cellc v_etotal v_ke v_pe v_epair off 2 \
file trajectory_npt_3_2.seamm_trj
run 500000
Although the two inputs shown above are quite different from each other, it would take a considerable amount of time for a non-expert user to precisely understand the details of each calculation and to be able to control the parameters in order to achieve the best possible results.
SEAMM, on the other hand, adopts a different strategy to expose all control parameters to the users through its user-friendly GUI. As such, the end-users can conveniently access or modify all software-specific variables in a flexible and interactive way with the default values already set to reasonable values.
Let us take a glance at the SEAMM GUI dialog for setting up a NPT calculation in LAMMPS package
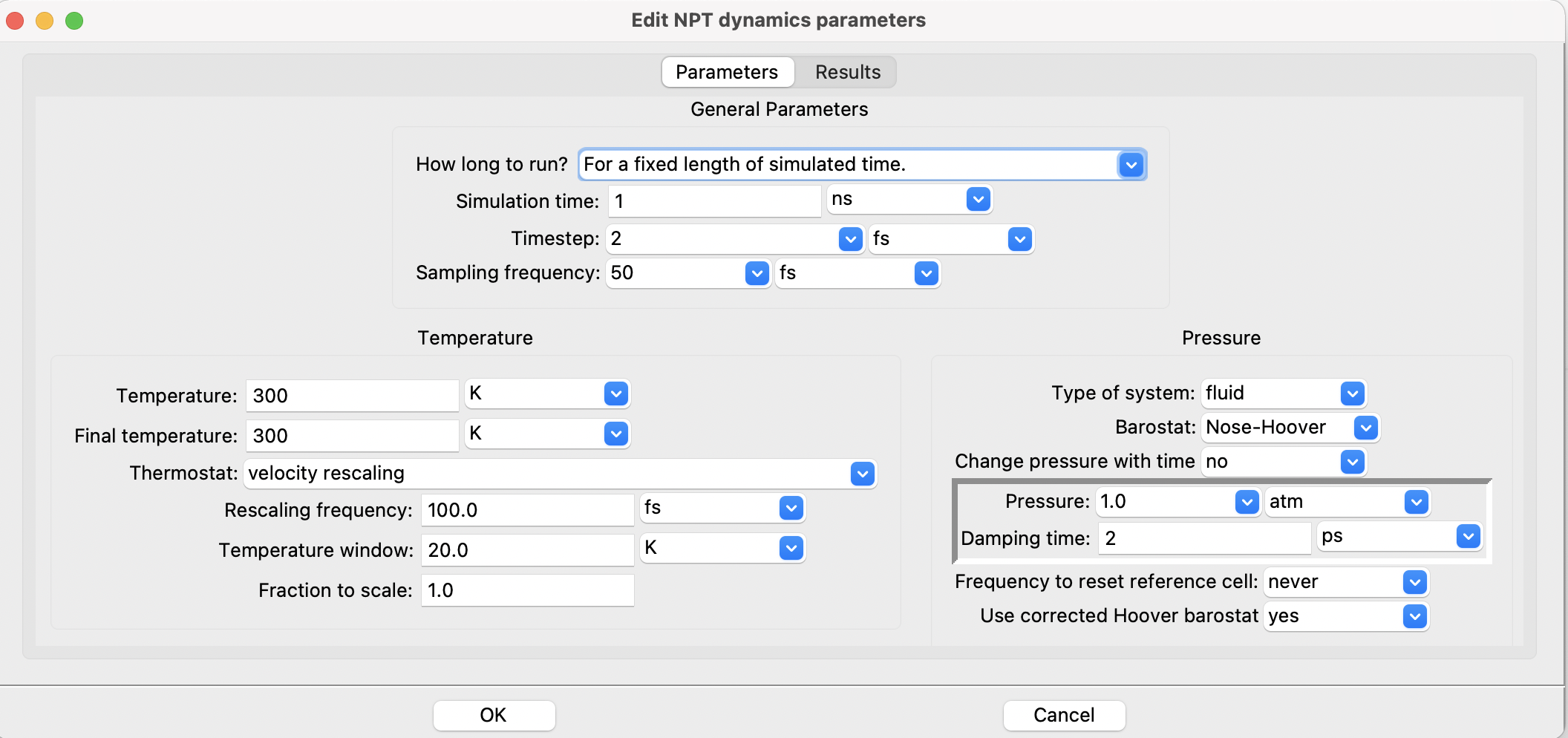
The LAMMPS NPT dialog#
The dialog provides a much easier to understand medium compared with the manual
modification of the input script files for each particular software. In a sense, the
GUI removes the need for learning various software-specific syntaxes and keywords
and replaces them with a user-friendly dialogue in a unified representation.
Let us consider the following LAMMPS dialogue and click on the Thermostat field:
The GUI then presents a list of possible choices:
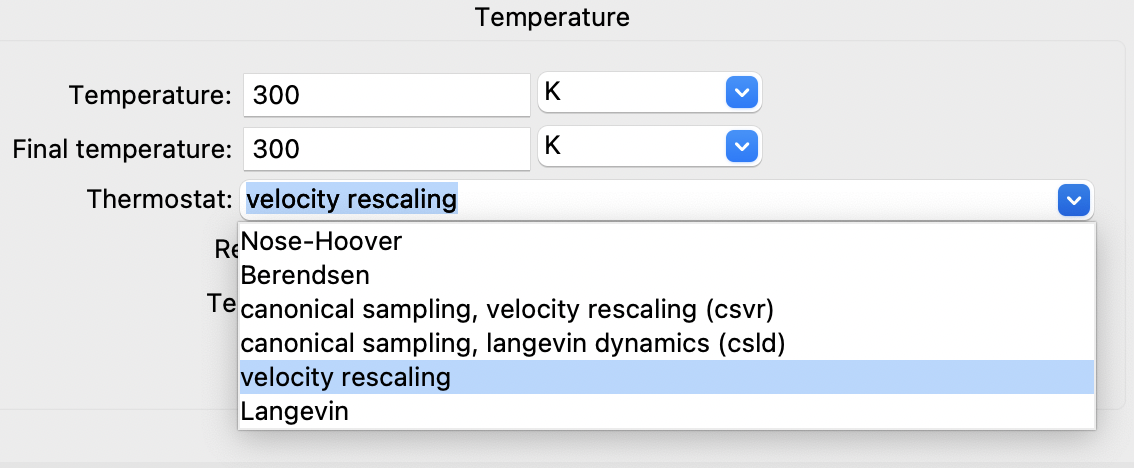
Choices for controlling the temperature in LAMMPS#
If we choose Berendsen's method for controlling the temperature, the dialog
automatically reconfigures to only show the pertinent options:
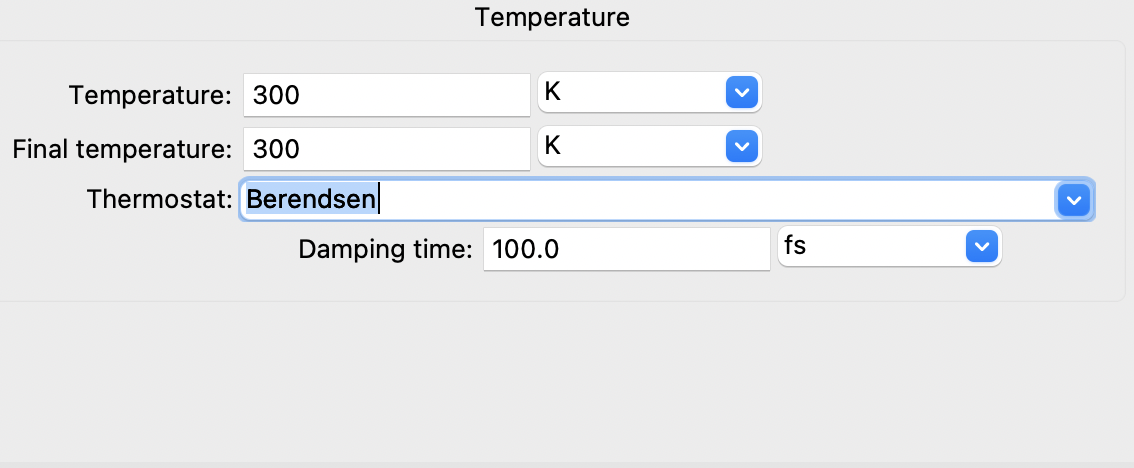
The control parameters for Berendsen’s method#
As such, while the GUI compiles and appropriate list of possible choices for the users, it pre-initializes each variable to a reasonable default value.
Similar to the LAMMPS dialogue demonstrated above, the GUI for GROMACS and other molecular dynamics packages can also be treated in the same user-friendly fashion within the GUI. While there might be some differences due to details of the software package implementation on the backend, most of the dialog would look the same.
The GUI is one of the most powerful tools that SEAMM offers to its users. While it is designed to remove the need for dealing with different software-specific syntaxes and keywords in the input scripts, it also exposes all functionalities of the simulation engine to the users in a unified, flexible, interactive and user-friendly interface.