
Atom Types#
One area not mentioned in our example OPLS paper is how to get the right parameters in each of the terms in the functional form. We glossed over this in the description of the section on the Functional Form, noting that we had various sets of parameters for the Lennard-Jones potential and for the torsions but not going deeper. If you read the paper you will notice that they use specific molecules and functional groups in the description of the parameters. In Table III the Lennard-Jones are described by the type of carbon atom and an example:
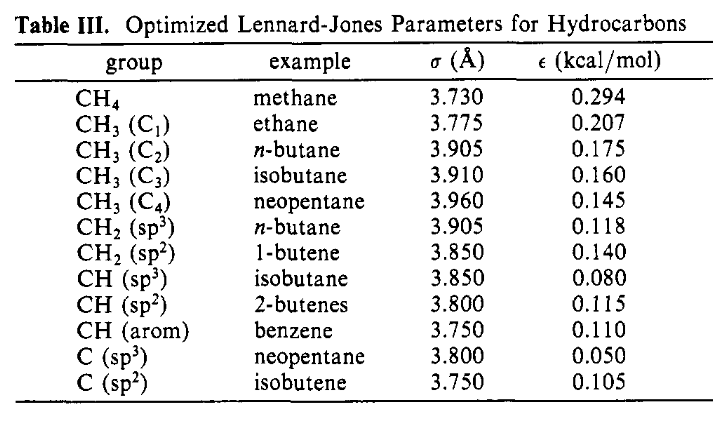
Figure 1: The Intermolecular Lennard-Jones Parameters#
The torsion parameters just use an example:
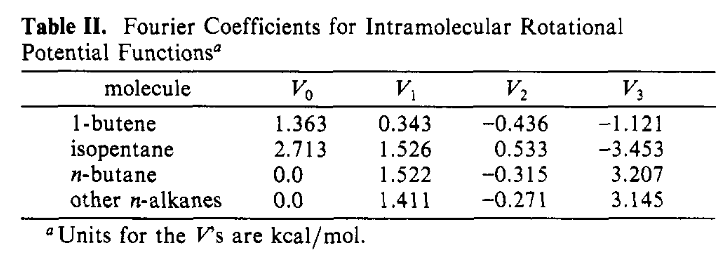
Figure 2: The parameters for the torsions.#
Presumably when this paper was written and computers were much smaller, the simulations were smaller and simpler so it was not an issue to set them up by hand, matching the atoms in the molecules to the descriptions in the paper. However as the systems became larger and more complicated, many of the forcefields for organic and biomolecules migrated to a more formal concept of “atom types”. The concept is simple: every atom has a local environment, which to a first approximation is defined by the nearby atoms and the bonding. If the atoms have identical environments, then they are the same “type”. If the environments are different but quite similar then the forcefield developer may lump them together in a single type, though what is similar is a matter of opinion, so different forcefields and even different versions of a forcefield may group atoms together in different way.
Working through an example using OPLS might help make this more clear.
isopentane#
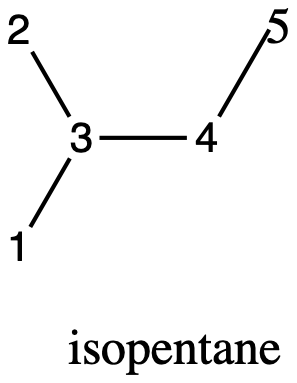
Figure 3: isopentane with the atoms numbered.#
The structure for isopentane has four different atom environments. Atoms 1 & 2 are topologically identical. The other methyl group, atom 5, is quite similar but not quite the same. The remaining two atoms, 3 and 4, are a >CH- and -CH2- group repectively.
Now the task is to decide which Lennard-Jones and torsional parameters are appropriate for this molecule. Starting with the Lennard-Jones parameters first, let’s examine the three methyl groups. OPLS has parameters for 12 different types of carbon groups:
CH4 as in methane
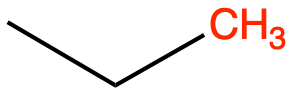
CH3 (C1) as in ethane
CH3 (C2) as in n-butane
CH3 (C3) as in isobutane
CH3 (C4) as in neopentane
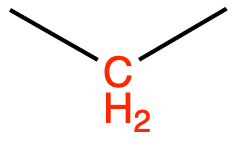
CH2 (sp3) as in n-butane
CH2 (sp2) as in 1-butene
CH (sp3) as in isobutane
CH (sp2) as in 2-butene
CH (arom) as in benzene
C (sp3) as in neopentane
C (sp2) as in isobutene
The two identical methyl groups, 1 & 2, are similar to the methyl groups in isobutane in that they are bonded to a tertiary carbon, so 4: CH3 (C3) is a reasonable choice for them. The other methyl group, 5, is probably closer to those in n-butane since it is bonded to a -CH2- group. Thus 3: CH3 - (C2) is probably the best choice.
Moving on to atom 4, the -CH2- group, there is only one possibility, 6: CH2 (sp3) as in n-butane. It seems to be a reasonable choice.
Finally, atom 3, the >CH- group, again has only one possibility, 8: CH (sp3) as in isobutane, and again this seems like a reasonable choice.
We can sum this up with the following picture, showing isopentane labeled by the atom types 1-12:
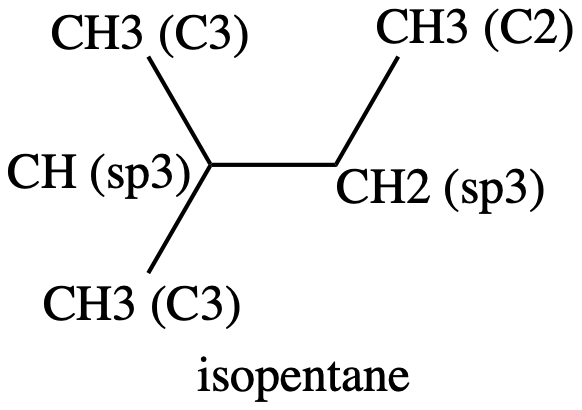
Figure 4: isopentane showing the atom types#
Now for the torsion types. There is only one “rotatable” bond in neopentane, that between atoms 3 and 4 in Figure 1, meaning that the angle between the planes defined by atom 2-3-4 and 3-4-5 changes the structure in a meaninful way. Not coincidentally of the three specific sets of torsional parameters in Table II above, one is noted as for neopentane so is clearly the one to use. Looking at the atom types in Figure 2, we could write this torsion as having the following atom types:
CH3(C3) - CH(sp3) - CH2(sp3) - CH3(C2)
Expressing Torsions in Terms of Atom Types#
Let’s express the structures for the different torsions in Table II as atom types an see what it looks like.
1-butene#
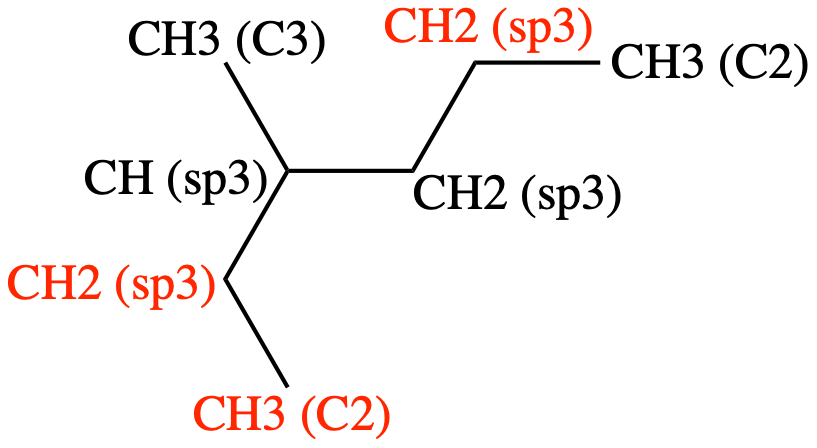
Figure 5: 1-butene showing the atom types#
The atom types labeling the atoms are reasonable given the information in Table 3. The torsion is labeled as follows:
CH2(sp2) - CH(sp2) - CH2(sp3) - CH3(C2)
n-butane#
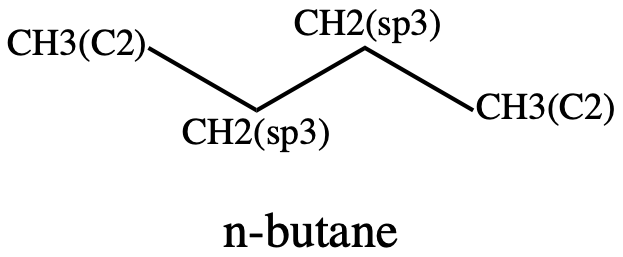
Figure 6: n-butane showing the atom types#
The torsion is labeled as follows:
CH3(C2) - CH2(sp3) - CH2(sp3) - CH3(C2)
n-pentane#
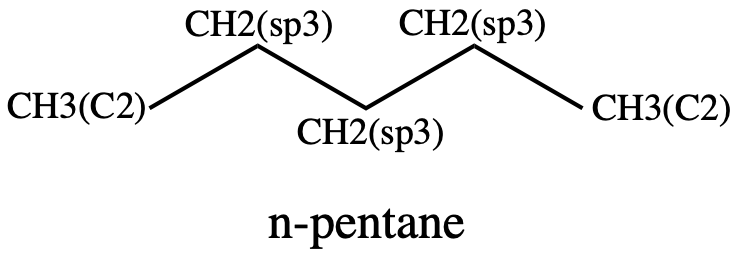
Figure 7: n-pentane showing the atom types#
The torsion is labeled as follows:
CH3(C2) - CH2(sp3) - CH2(sp3) - CH2(sp3)
There is a second torsion in this molecule, starting one atom into the chain:
CH2(sp3) - CH2(sp3) - CH2(sp3) - CH3(c2)
Note however that this is the same as the first torsion except with the atoms ordered in the reverse direction. Which direction you look down the rotating bond makes no difference, so these two are identical.
For longer normal alkanes this torsion would appear at both ends. In the middle of the chains, all four atoms involved in the torsion would be -CH2- groups:
CH2(sp3) - CH2(sp3) - CH2(sp3) - CH2(sp3)
In Table II the last torsion is labeled as for all other n-alkanes, so in OPLS both of these torsions, which are just slightly different, are treated identically.
Putting it all together#
We have seen how atoms in similar environments can be grouped into atom types. This allows us to recast the OPLS forcefield in terms of atom types, and associate the parameters in terms of atom types.
The following table expands their Table III giving a picture defining the atom type. Red indicates the atom whose type is described, with the picture giving a substructure, meaning that terminal atoms may be part of a longer chain or have other substituents unle a full valence is indicated explicitly.
Atom Type |
Picture |
\(sigma\) (Å) |
\(epsilon\) (kcal/mol) |
|---|---|---|---|
CH4 |
|
3.730 |
0.294 |
CH3(C1) |
|
3.775 |
0.207 |
CH3(C2) |
|
3.905 |
0.175 |
CH3(C3) |
|
3.910 |
0.160 |
CH3(C4) |
|
3.960 |
0.145 |
CH2(sp3) |
|
3.905 |
0.118 |
CH2(sp2) |
|
3.850 |
0.140 |
CH(sp3) |
|
3.850 |
0.080 |
CH(sp2) |
|
3.800 |
0.115 |
CH(arom) |
|
3.750 |
0.110 |
C(sp3) |
|
3.800 |
0.050 |
C(sp2) |
|
3.750 |
0.105 |










And the torsions from Table II, using the atom types as labels:
Torsion |
V0 |
V1 |
V2 |
V3 |
|---|---|---|---|---|
|
1.363 |
0.343 |
-0.436 |
-1.121 |
|
2.713 |
1.526 |
0.533 |
-3.453 |
|
0.0 |
1.522 |
-0.315 |
3.207 |
|
0.0 |
1.411 |
-0.271 |
3.145 |
Automatic Assignment#
As computers have gotten more and more powerful the complexity of simulations has grown by leaps and bounds. It is no longer viable to assign the atom types to the structure by hand, nor would that lead to reproducible simulations since there is often a choice of similar parameters, so different people will choose different options.
There are two approaches in general use for automatically assigning the atom types and hence the parameters for a simulation. Perhaps the initial approach was to use template or residue libraries, relying on the regular structure and naming of proteins. A later paper of the extension of OPLS for proteins illustrates the approach:
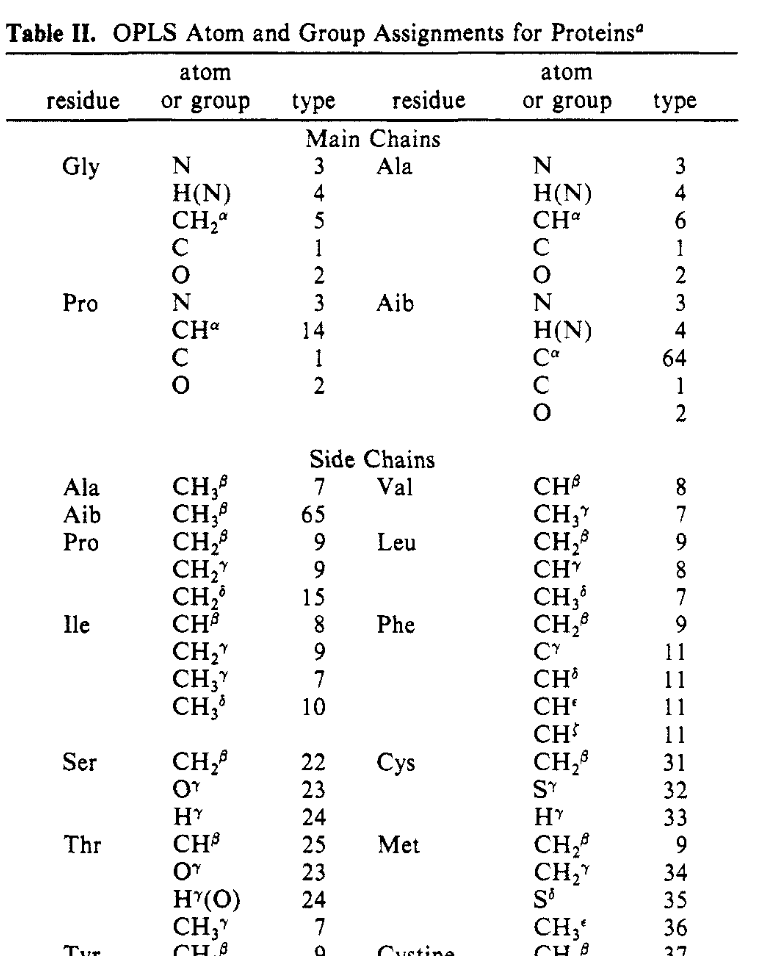
Figure 8: Atom type assignment for proteins in OPLS.#
This table gives the residue, standard atom name, and atom type for the standard amino acids. OPLS has migrated to using numbers for the atom types, compared to the more descriptive text used in the original paper. CH3(C2) has become atom type 10 and CH2(sp3), 9. The nonbond parameters remain unchanged from the original paper, just the name has changed.
The template approach is certainly simple, and has been and still is widely used for biomolecules – peptides, proteins, DNA and lipids – where there are strong naming conventions for the atomms.
However, for more general organic chemistry templates are not useful since there is no standard naming conventions for the atom. This gave rise to the second approach, some form of chemical perception. With some thought and effort, it is possible to translate the pictures above into e.g. SMARTS for substructure searches for the atom type assignment. This approach was certainly implemented for the CVFF and CFF series of forcefields at Biosym in the early 1990’s, and with the widespread adoption of SMARTS and similar tools for substructure matching is becoming more feasible and widespread.
Summary#
Atom types provide an approach for collecting atoms in simlar local environments into groups, and specifying which parameters to use for the terms in a forcefield. Typical valence forcefields have terms relying on one, two, three and four atoms – the nonbonded terms, bonds, angles, and torsions and out-of-plane terms, respectively. For a given molecule or system the atom types are assigned, either by hand, using template libraries, or by chemical perception. Once the atom types are assigned to the structure finding the correct parameters for each term is reduced to a lookup in tables indexed by the atom types.