
1: A First Calculation#
Note
The flowchart for this tutorial is available at Zenodo and also can be directly loaded into SEAMM by opening from Zenodo. The job for this tutorial is available on MolSSI’s public server, Job 423.
Introduction#
This tutorial mirrors the first tutorial in the FHI-aims tutorial series, doing a similar calculation in SEAMM. We’ll create a water molecule and optimize the structure using FHI-aims.
Note
This tutorial is almost identical to the first SEAMM tutorial but uses FHI-aims instead of DFTB+, and uses a different molecule – water rather than ethanethiol. The steps are the same, but the some of the details are different. Since this is the first tutorial, we’ll go through the steps in detail.
To get started, either double-click on the SEAMM app if you installed it, or open a terminal and activate the seamm conda environment and start SEAMM:
conda activate seamm
seamm
This will bring up a window like the one shown below:
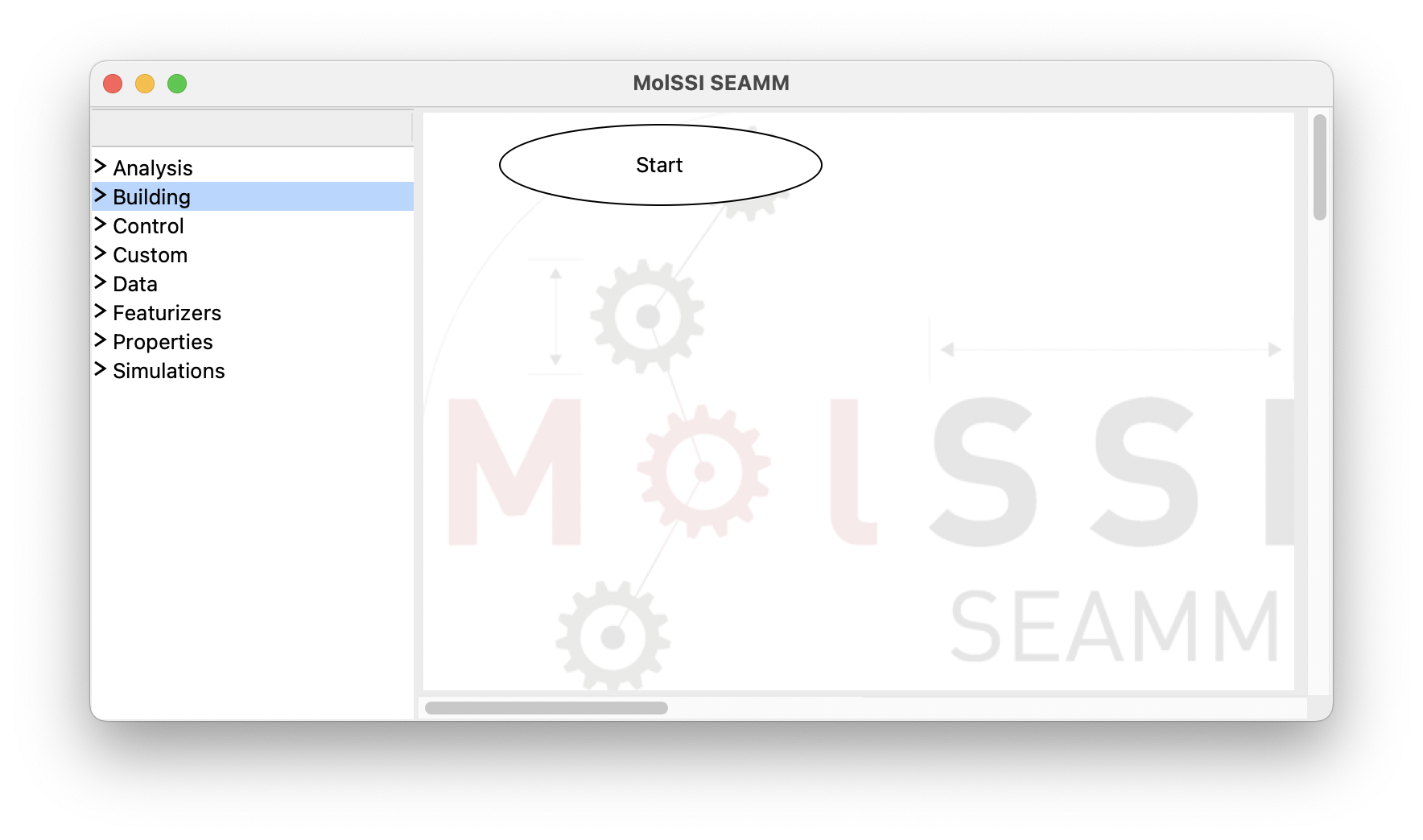
SEAMM initial window#
The window consists of two panes separated by a sash – the grey line. If you hover over the sash the cursor will change to a horizontal double-pointed arrow enabling the adjustment of the pane sizes. The left pane contains the list of available plug-in categories. Clicking on each category expands its content and represents it as a list of available plug-ins. Clicking on the same point on the screen will hide the expanded list:
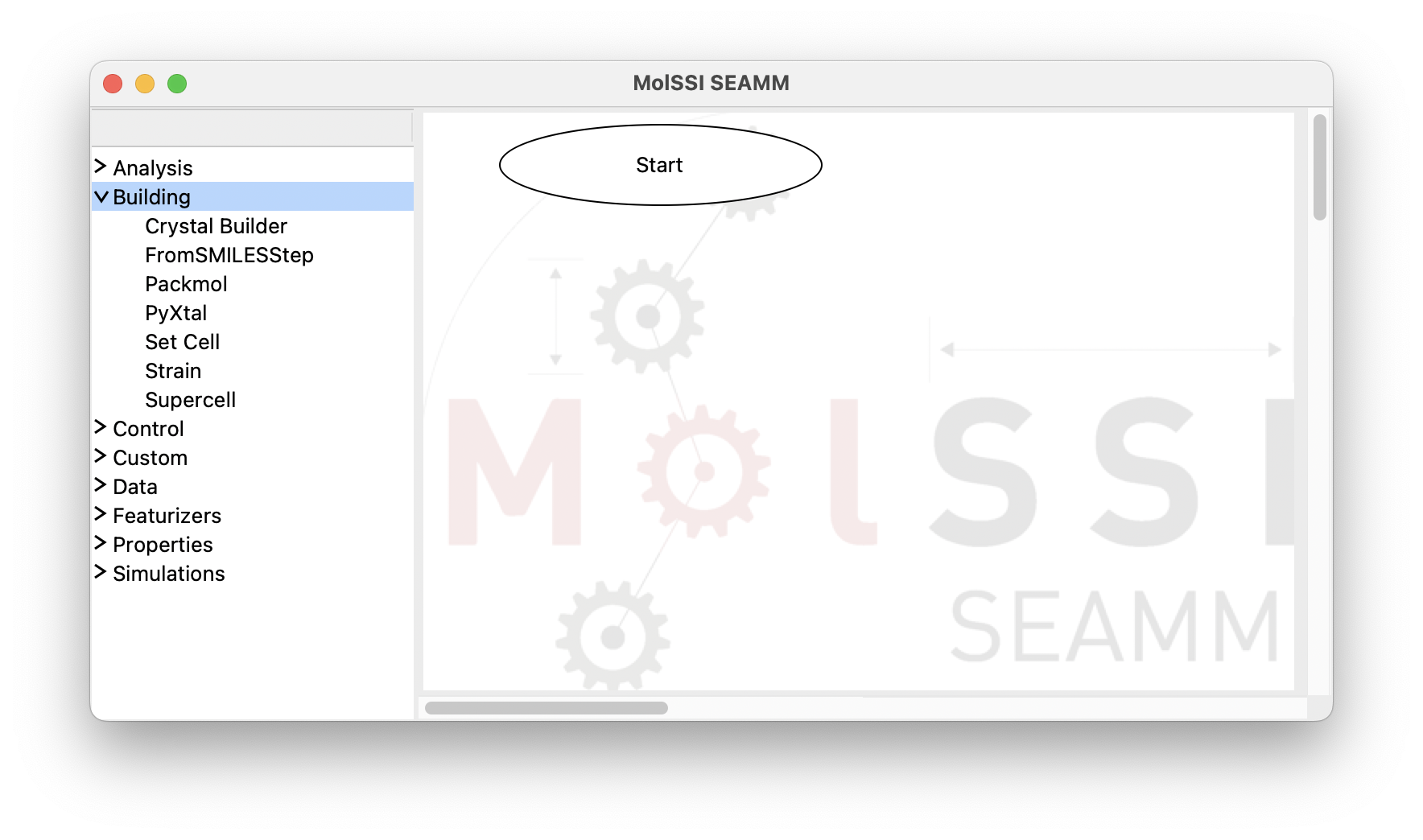
The SEAMM window with the Building category of plug-ins expanded#
The right-hand pane contains the initial step of a flowchart, Start. There must be one
and only one start step in a flowchart because this is where execution starts. Users
cannot delete or add a Start step.
Creating the Flowchart#
Let us create a simple flowchart and add a molecule to it using SMILES. SMILES is a specific representation of a chemical structures in the string format. Then, we add a new step, in which we optimize the geometry of the molecule, imported in the previous step, with FHI-aims.
By clicking on the FromSmilesStep from the Building section, a rectangular box
for the new step labeled as from SMILES appears in the right pane. This box is also
connected to the Start step:
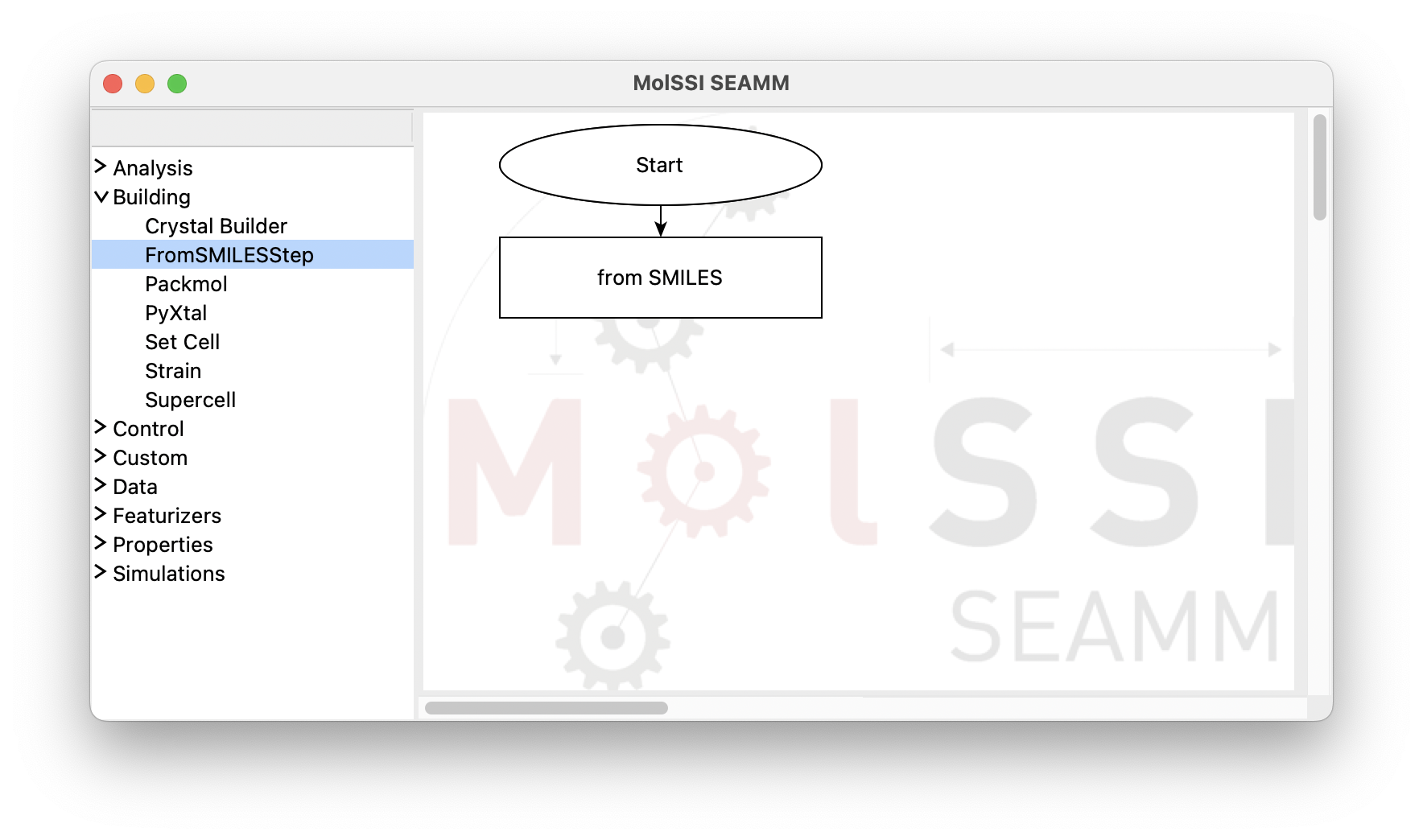
The flowchart after adding the FromSMILESStep#
If you have accidentally clicked twice on the FromSmilesStep or any other menu item
that you did not want, just right-click on the unwanted step and select delete from
the pop-up menu:
Deleting a step from a flowchart#
In the next stage, we need to set the SMILES string variable for the added
molecule. Almost every step has a dialog window that allows the users to set parameters
and control the details of the task in each step of the workflow. In order to access the
dialog, one can either right-click on the step box and select Edit... from the menu
or just double-click on the step box to open it:
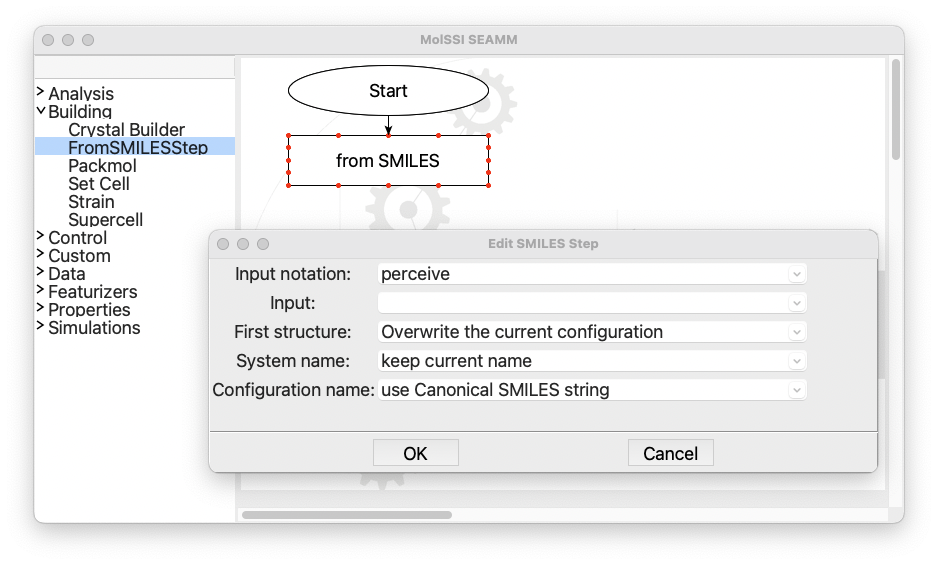
The dialog for the FromSMILES step#
Click in the field labeled Input: and type O, which is the SMILES representation
for water. Leave everything else as is and click on the OK button:
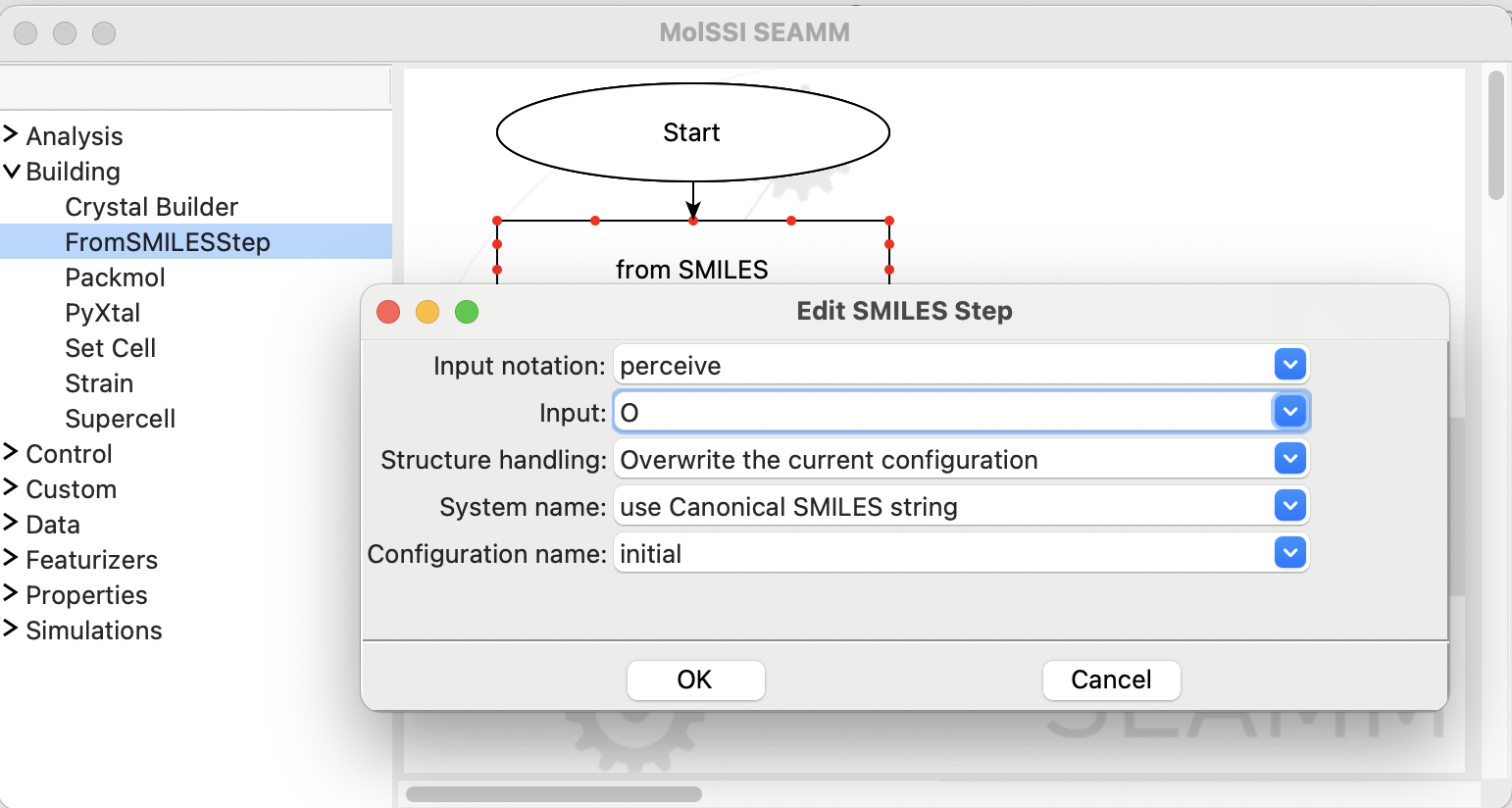
The completed dialog for the FromSMILES step#
Note
While the name FromSMILES suggests that you need to enter SMILES, it has been
enhanced over time to also handle chemical names, InChI, and InChIKeys. The first
pulldown allows you to specifiy which you are entering; however, perceive will
typically correctly understand the text that you typed in.
You can also change how to store the new structure and the name of the system and configuration it is stored in, but for now just use the defaults.
Now, open the Simulations section in the left panel and add a FHI-aims step:
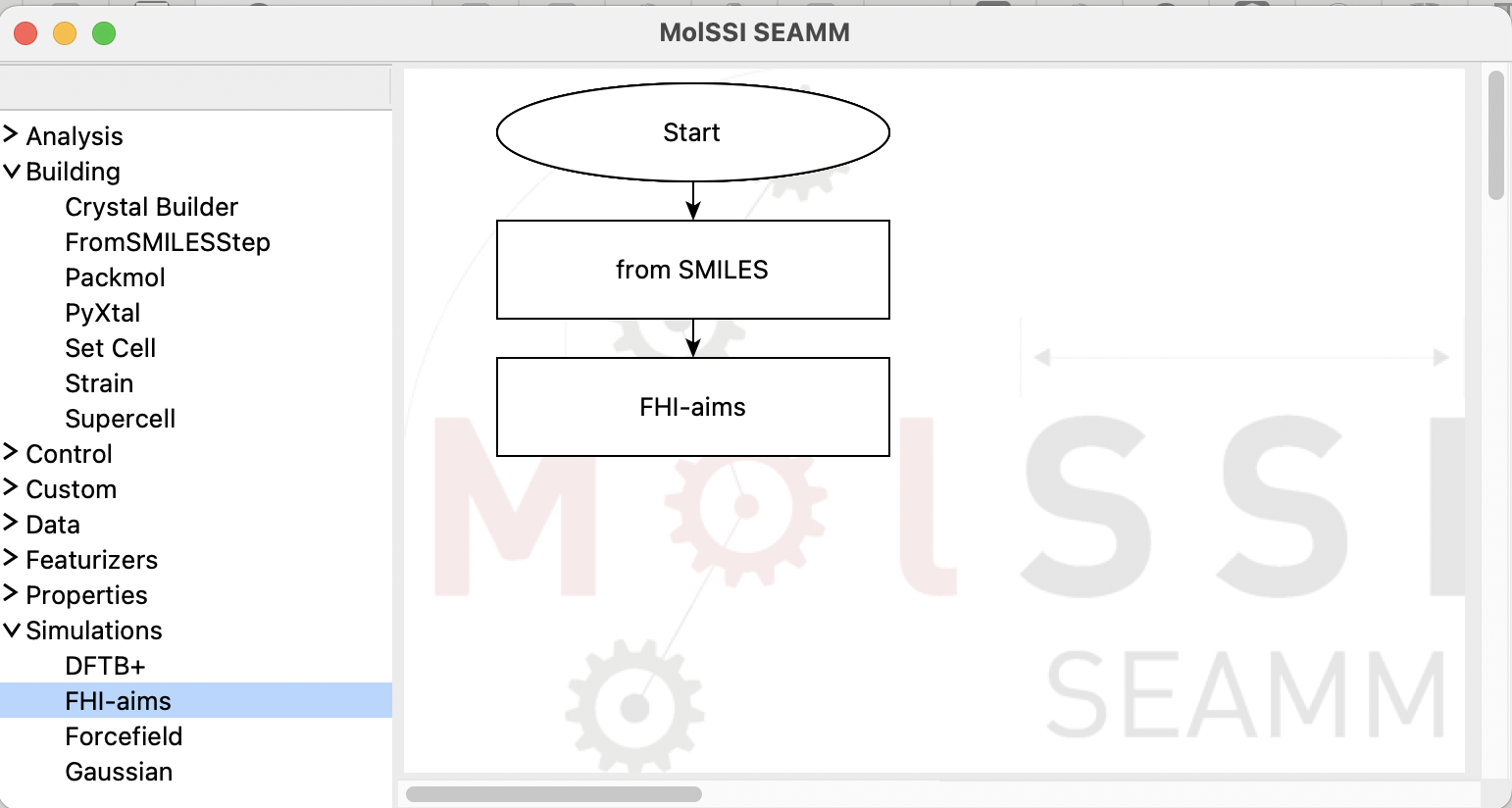
The flowchart with the FHI-aims step added#
Then, open the dialog for the FHI-aims step:
The dialog for the FHI-aims step#
Note that this dialog looks like the main SEAMM window. Some steps, like the FromSMILES
step, perform one task and thus, can be handled by a single dialog. However, many
simulation codes such as FHI-aims are capable of performing multiple tasks and often have
their own syntax.
Open the Simulation section on the left panel and add the Optimization step to
the subflowchart:
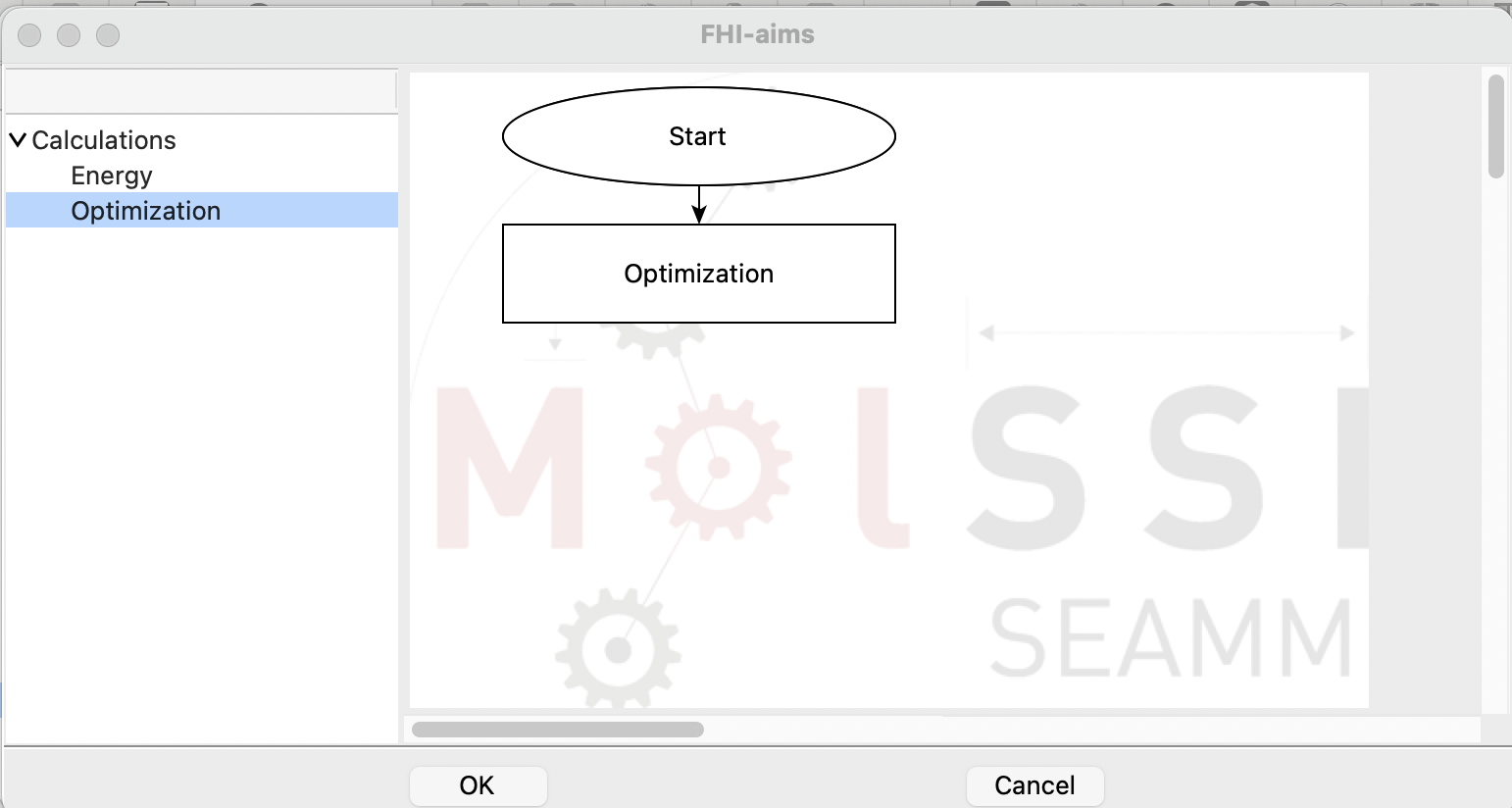
The subflowchart for the FHI-aims step#
Open the Optimization dialog:
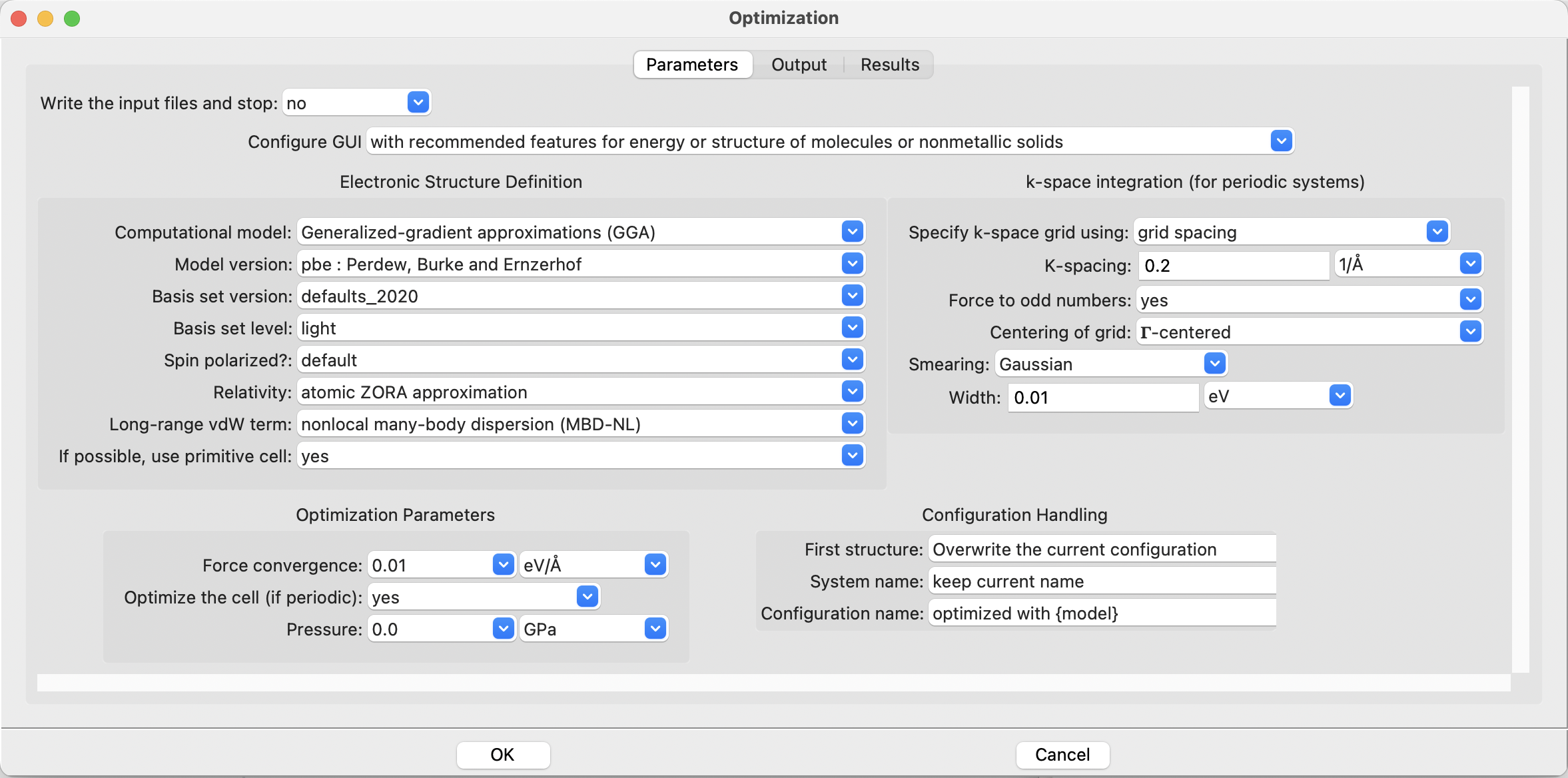
The FHI-aims Optimization dialog#
There are a number of control paramters available. On the left side of the dialog, there are options for controlling the Hamiltonian and the physical approximations being made. The right hand panel shows the options for the numerical integration over the Brillouin zone for periodic systems. These options are ignored for molecular systems. The bottom part of the dialog consists of parameters for controlling the geometry optimization procedure.
SEAMM strives to have reasonable default values, so can start with the defaults, which actually correspond almost exactly to the input specified in the FHI-aims tutorial for water. The only difference is that by default the FHI-aims plug-in adds long-range van der Waals terms. They will not affect the optimization of water, but you can turn them off if you want to have an identical calculation to the generic FHI-aims tutorial.
Note
FHI-aims and several other codes in SEAMM can handle both molecular and periodic systems, which is why you see options for using the primitive cell, the k-space integration grid, and whether to optimize the cell. These are ignored for molecular systems – there is no cell – but allow you to create flowcharts that can work with both molecular and periodic systems.
Before leaving the dialog, click on the Output tab at the top of the dialog:
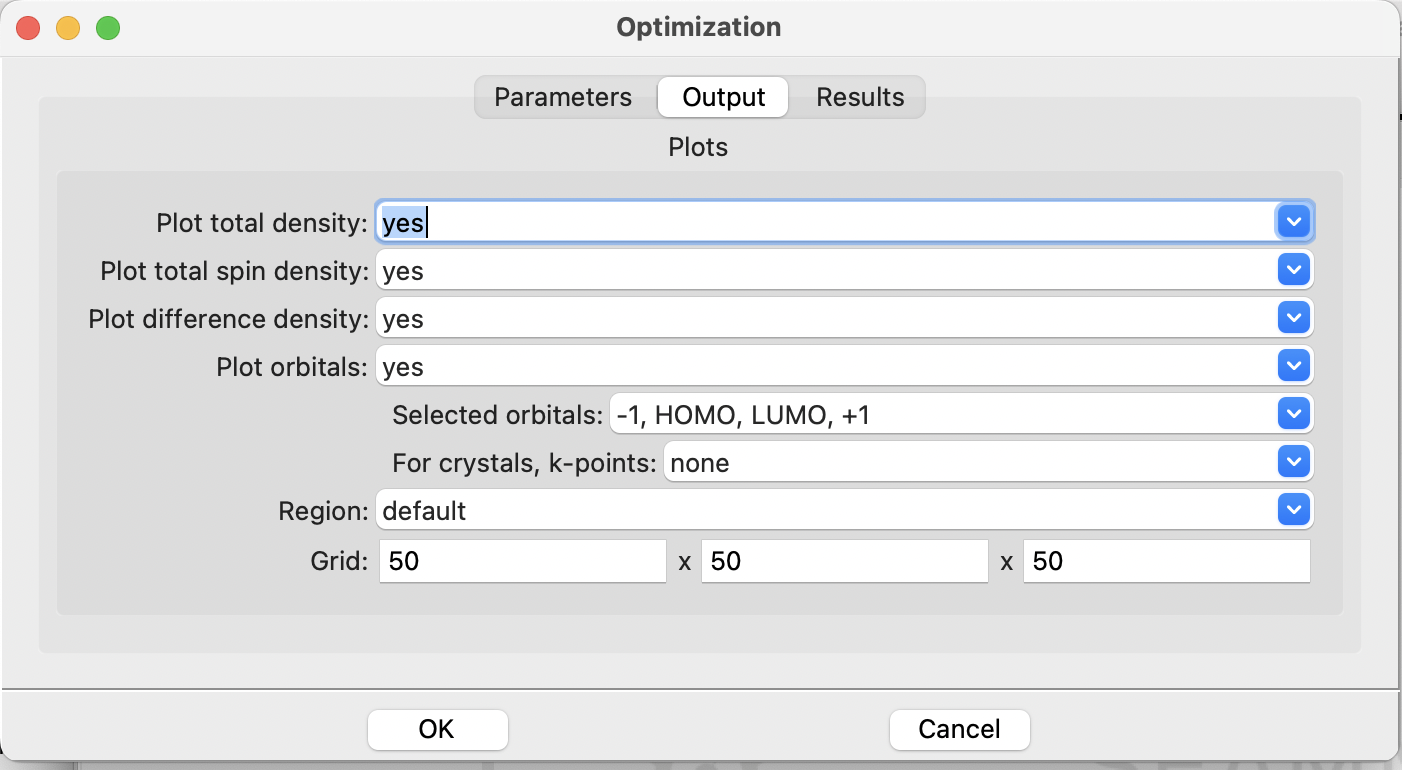
The FHI-aims Optimization dialog, output tab#
This is where you control plotting the total charge density, spin-density (for
spin-polarized calculations), and orbitals. By default it does not plot any densities or
orbitals. Change the items to yes, leaving the selected orbitals -1, HOMO, LUMO,
+1 which asks the code to plot the HOMO and LUMO orbitals as well as the next orbital
below the HOMO (-1) and the next orbital above the LUMO (+1). Again you see some
controls for periodic systems, which you can ignore. The defaults are fine, so click
Cancel to close the dialog with no changes.
Tip
If you do not intend to make any changes in a dialog, it is a good idea to close it with
the Cancel button. It is a common habit to click OK, but pressing OK
saves any changes including ones you made accidentally by e.g. looking at the options
in a pulldown. Such a mistake can lead to a calculation that you didn’t intend, with
puzzling results. We recommend using the Cancel button in any dialog in SEAMM or
any other program unless you actually want to make changes. It is a good habit to get
into.
Click OK to close the dialog to save your changes.
Running the Job#
To submit the flowchart as a Job, click on the File menu and select Run or use
the accelerator (⌘R on a Mac, ^R on Windows or Linux) to get the following dialog:
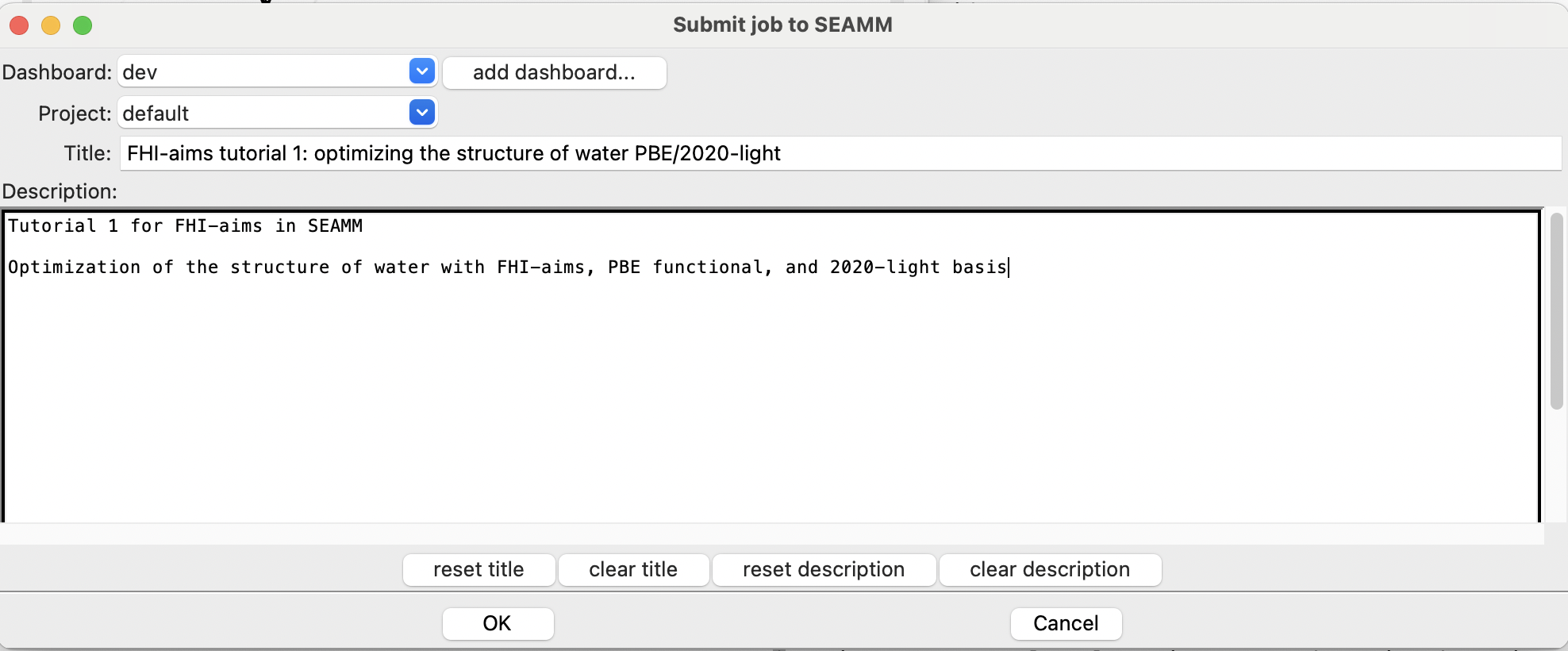
The Run dialog in SEAMM#
Note
The very first time you run, you will need to provide a username and password for the Dashboard. If so, this dialog will appear first:
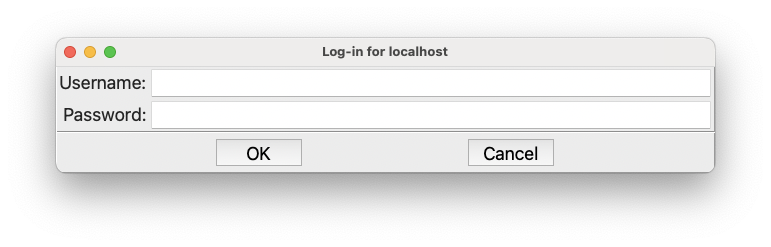
The login information for the Dashboard#
If you don’t know your username and password go to Managing the Dashboard and then come back here.
Add a useful title in the Title field. Briefly describe your goal and the details of
your simulation workflow within the large text area at the bottom of the
dialog. Finally, click OK to execute the calculation.
As noted at the beginning this tutorial you can download the flowchart from Zenodo and view the results of the calculation in the public MolSSI Dashboard at Job 423.
The second SEAMM tutorial will show you how to look at your job and its results in the Dashboard.
Topics Covered#
Adding steps to a flowchart.
Editing the parameters for the steps.
Submitting a job.