
Creating a Dataset#
This section covers how to create a dataset in QCArchive and add entries to it. With minor changes this is also how you add entries to an existing dataset, so both are covered here.
- The steps are straightforward
Create the dataset in QCArchive
Get one or more molecules into SEAMM
Upload them to the dataset
This flowchart is a very simple example of doing
this.
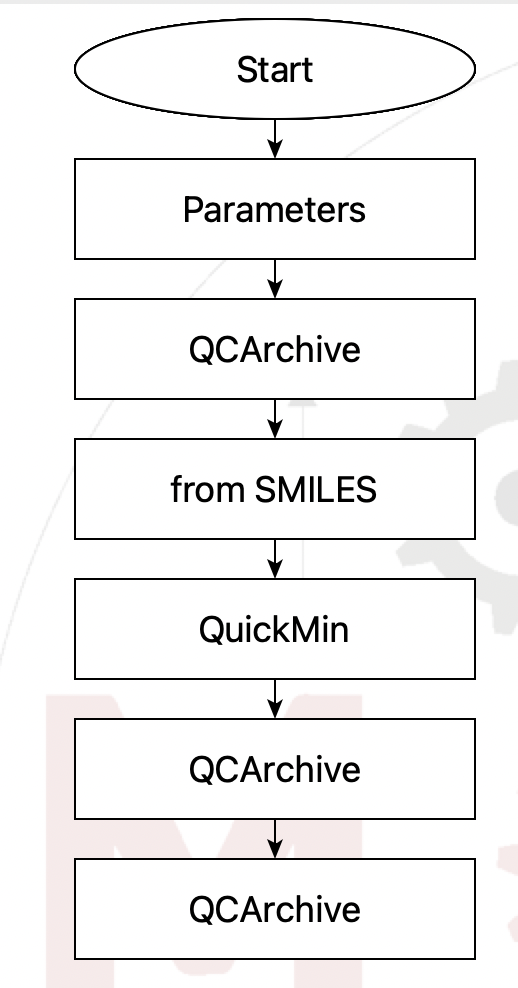
Flowchart to create a QCArchive dataset#
- The steps are
Set up a parameter to get the SMILES string
Create the dataset in QCArchive if it doesn’t exist
Create the structure from SMILES
Quickly optimize the structure with a forcefield to make it reasonable
Add the structure to QCArchive as an entry in the dataset
List the entries in the dataset to check that it worked
Obviously this a toy example. In reality you would want to impoprt a number of structures, probably from files, using a loop. But this simple flowchart captures the essence of the problem.
The first QCArchive step is set up like this
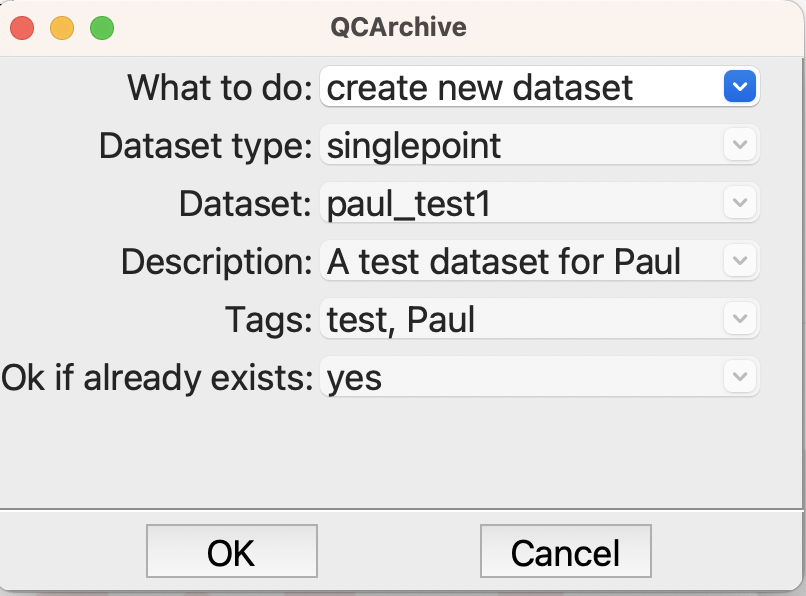
Options for creating a QCArchive dataset#
Hopfully this is straightforward and easy to understand. The interesting option is the last, saying that it is OK if the dataset exists. The first time we run this it will create the singlepoint dataset paul_test1 in QCArchive. Subsequently, it won’t do anything becasue the dataset exists, so we can run this flowchart a number of times to add several molecules.
The next couple of steps in the flowchart creates the structure from the SMILES string that the initial Parameter step captured, and then optimizes it with a forcefield in order to clean the structure.
The second QCArchive set adds the structure to the dataset
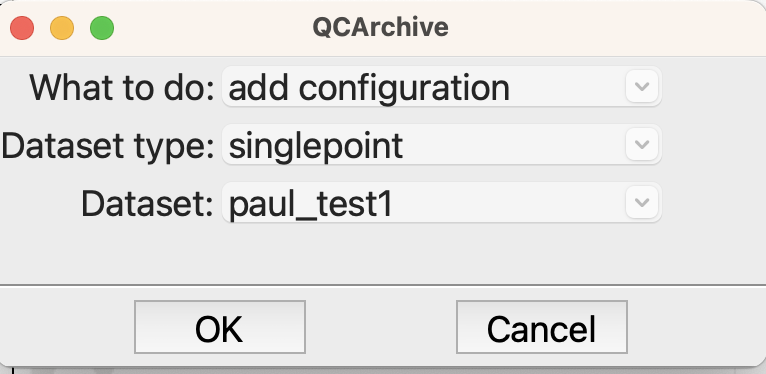
Add the structure to the QCArchive dataset#
If all goes according to plane, the last QCArchive step will list the entries in the dataset so that we can check that the new structure has been added.
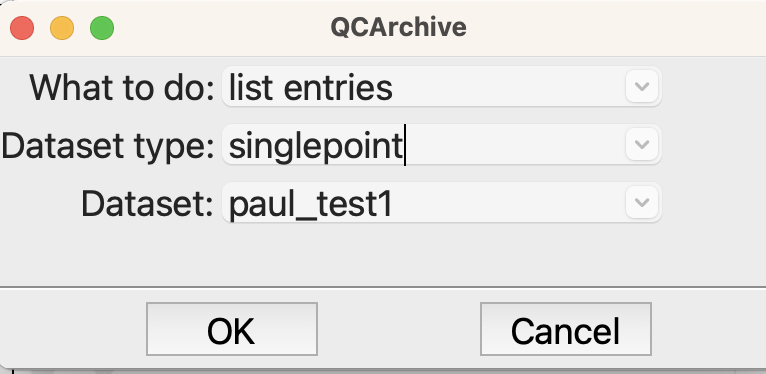
Options for listing the entries in the dataset#
The output from running the flowchart is:
Running the flowchart
---------------------
Step 0: Start 2023.4.8
Step 1: Parameters 2023.1.23
The following variables have been set from command-line arguments,
environment variables, a configuration file, (.ini), or a default value, in
that order.
+------------+----------+-------------+-----------------------------+
| Variable | Value | Set From | Description |
+============+==========+=============+=============================+
| SMILES | c1ccccc1 | commandline | The SMILES for the molecule |
+------------+----------+-------------+-----------------------------+
Step 2: QCArchive 2023.3.28
Will create a new singlepoint dataset paul_test1 in the QCArchive
Validation Project Server.
Created singlepoint dataset paul_test1.
Step 3: from SMILES 2021.10.13
Create the structure from the SMILES 'c1ccccc1', overwriting the current
configuration. The name of the system will be the canonical SMILES of the
structure. The name of the configuration will be initial.
Created a molecular structure with 12 atoms.
System name = c1ccccc1
Configuration name = initial
Step 4: QuickMin 2023.1.14
Minimizing the structure with the best available forcefield, with a maximum
of 1000 steps. The optimized structure will be put in a new configuration
with 'optimized with ' as its name.
The minimization converged in 74 steps to 17.872 kJ/mol. The final structure was
saved in the new configuration named 'optimized with GAFF'.
Step 5: QCArchive 2023.3.28
Will add the current configuration to the singlepoint dataset paul_test1 in
the QCArchive Validation Project Server.
Added c1ccccc1/optimized with GAFF to the dataset.
Step 6: QCArchive 2023.3.28
Will list the entries in the singlepoint dataset paul_test1 in the QCArchive
Validation Project Server.
There are 1 entries in the singlepoint dataset paul_test1:
c1ccccc1/optimized with GAFF
That looks good! The last couple lines let us know that we added an entry for benzene to the dataset. If we run again with toluene as the molecule the last part looks like this:
There are 2 entries in the singlepoint dataset paul_test1:
Oc1ccccc1/optimized with GAFF
c1ccccc1/optimized with GAFF
which is correct: the dataset now has two entries, benzene and toluene.