
Recipe 1 extended: More flexible, useful workflows#
Introduction#
Recipe 1 is a great introduction to running DFTB+. If you read the recipe on the DFTB+ website, it is quite long and detailed because it is introducing the input to DFTB+ as well as the manually creating the structure. By contrast, setting up the same calculation in SEAMM, example is trivial – use SMILES to create the structure and a very simple DFTB+ flowchart to optimize the structure. The only thing we had to change from the defaults was the parameter set to use. Most of the tutorial was about understanding the output in the Dashboard, job.out in particular.
Hard-wiring the molecule in the from SMILES step works, but is a bit cumbersome. Sure, you can edit the flowchart every time you want to run a different molecule, but there should be a better way. There is! SEAMM supports input parameters for flowcharts. The parameters step handles this. You could edit the exisiting flowchart to add this, but let’s try something new. Let’s get the flowchart from Zenodo.
If you are not familiar with Zenodo you might want to head over there and look around. Zenod is a fantastic resource for storing and sharing data, documents, release of code, etc. getting a DOI in the process. DOI’s are used for archival storage. Once a DOI is minted for the data, the data can no longer be changed, though new versions, with new DOI’s can be stored. You can also go directly to the flowchart that we are interest in at https://doi.org/10.5281/zenodo.7013212 Zenodo also supports communities, and there is one for SEAMM flowcharts where you can search and browse available flowcharts. As you become more familiar with SEAMM you should consider uploading useful flowcharts to Zenodo and submitting them to the community. You can upload directly from SEAMM!
Tutorial#
You can also open a flowchart from Zenodo directly in SEAMM. Start SEAMM and either select File/Open or use the shortcut ctrl-O (cmd-O on a Mac). When the dialog appears, change the From filed from local files to Zenodo. The Show flowcharts where section, click the + button and fill out the line to read “must have any field contains SMILES” and click the Search button:
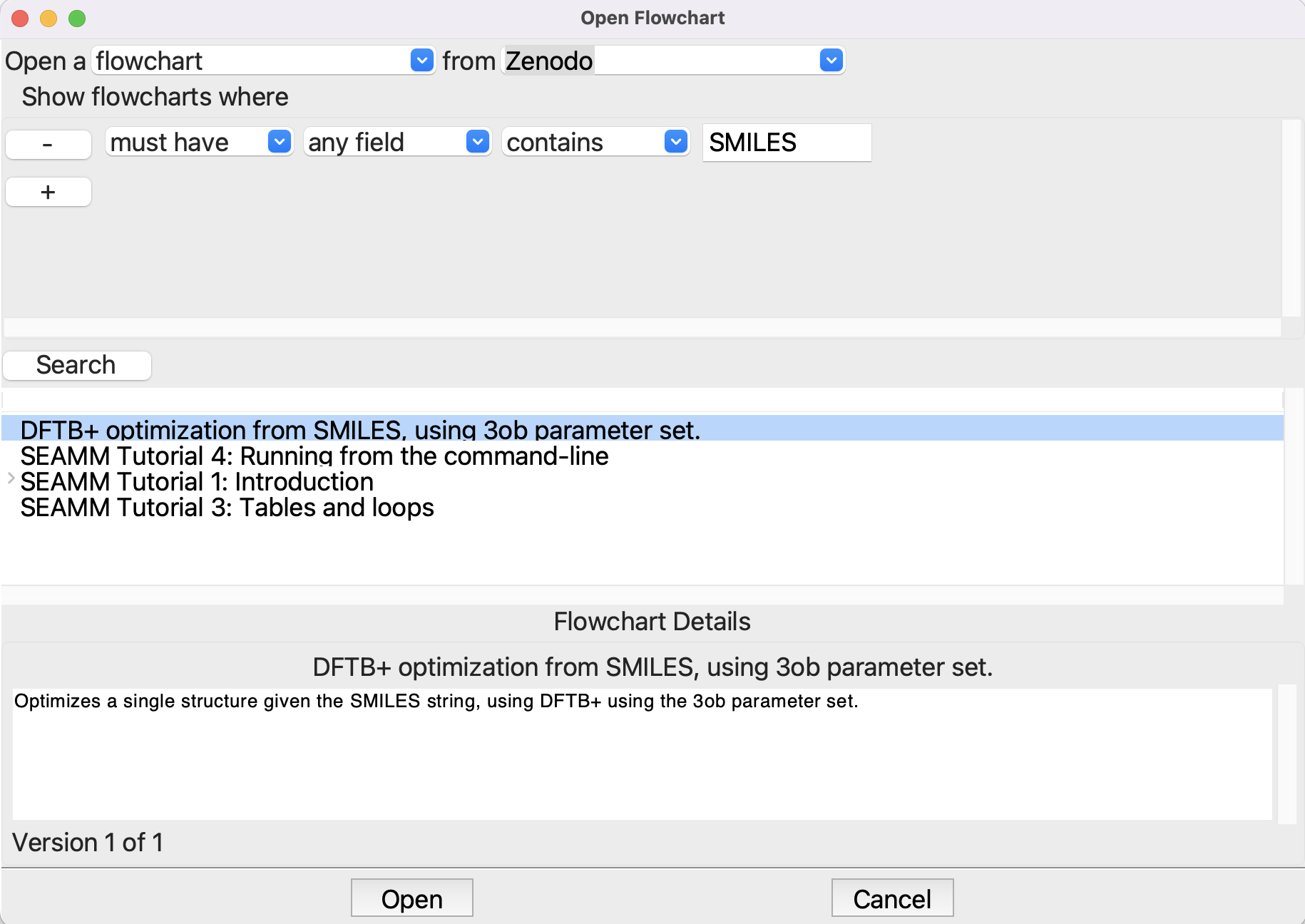
Opening a flowchart from Zenodo#
We want the flowchart “DFTB+ optimization from SMILES, using 3ob parameter set”, so select it. The description of the flowchart will appear in the Details area – this is how you can browse the flowcharts to find one that you want. Click Open to open the selected flowchart:
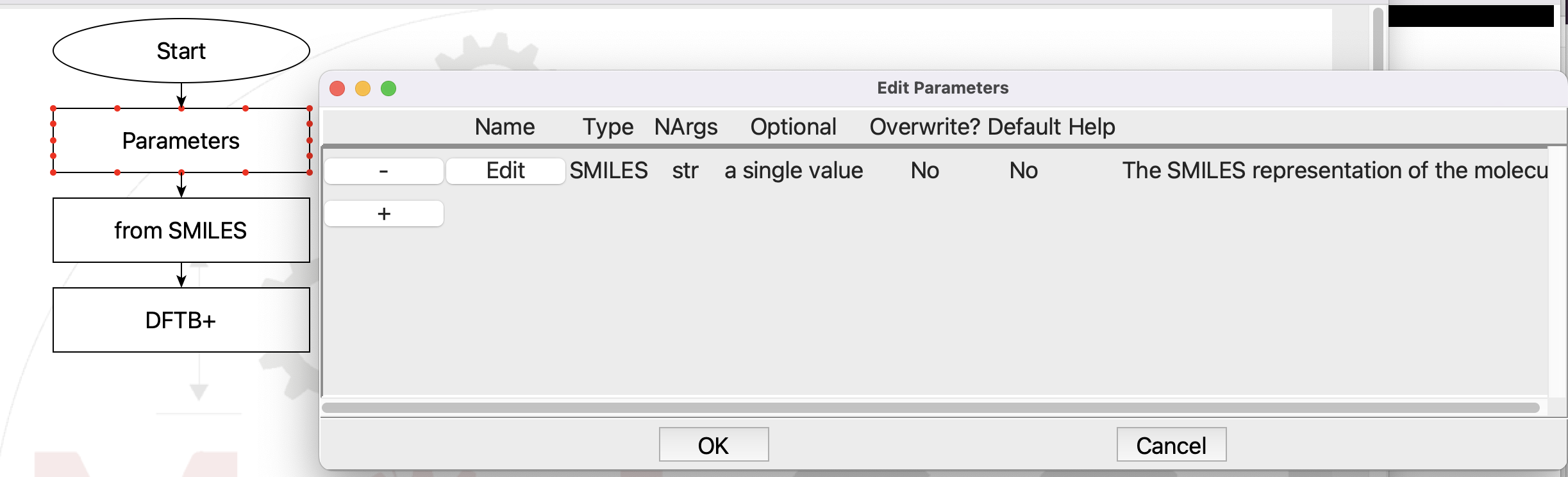
Flowchart with Parameters dialog open#
You can click the Edit button next to the SMILE parameter to see how the parameters are setup. Just click Cancel to exit the dialog so that you don’t accidentally make any changes. This defines a parameters SMILES that we set when we submit the job. This allows you to run different molecules easily. There is one more change from the original flowchart, in the from SMILES step:
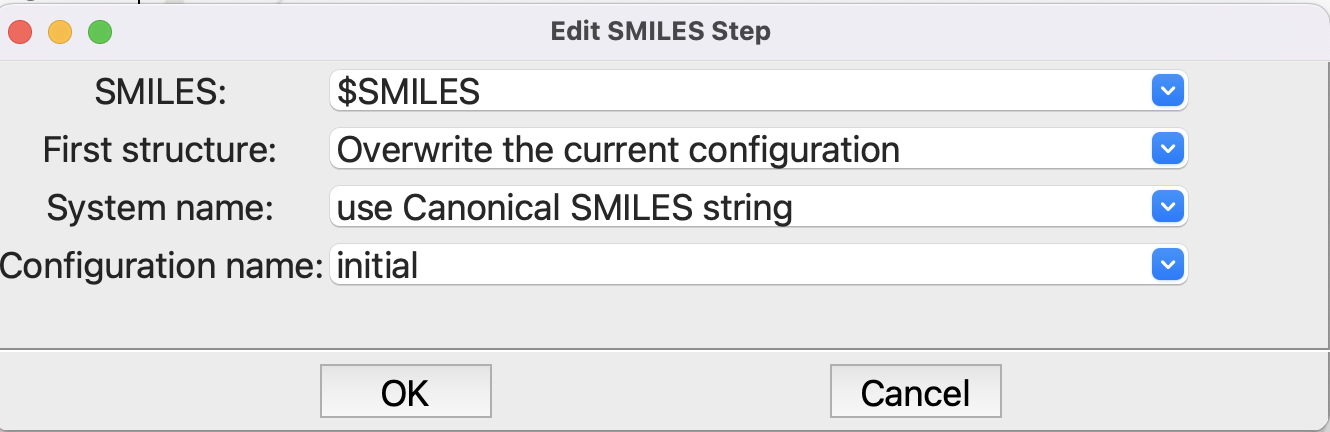
Editing the from SMILES step#
Note that rather the have the SMILES string in the first field, we use the parameter prefixed by a dollar sign to indicate that it is a variable, not a string.
That’s it! Now submit the job (File/Run or ctrl-R):
Submitting a job with a parameter#
Once the job finishes, you can look at the structure and outputs. Note that, unless you changed the flowchart, it is using the newer 3ob parameter set, so if you run water again you will get similar but slightly different results. Note that for toluene the calculated \(ΔE_f = 40\) kJ/mol. The experimental \(ΔH_f ≈ 50 ± 1\), so reasonable agreement.
Summary#
In this tutorial you have seen how to make a general, useful flowchart. SMILES is an excellent way to construct small organic molecules, and it is easy to find the SMILES for many molecules by searching on the Internet or looking in databases such as PubChem. The SMILES “language” is also quite easy to learn. With a little practice you can easily generate most small molecules. Later in these tutorials you will see a similar approach based on structure files, which is a bit more general and also works for crystalline systems, where there is no equivalent of SMILES.
You should also be aware of the limitations of this approach. The principal issue is the optimized structure is a local minimum, not necessarily the global minimum, which is usual what you are interested in. This is not an issue with small molecules, but as the system size increases, and particularly for “floppy” molecules with many rotatable bonds, finding the global minimum is quite difficult. And in some senses the concept of a structure – a minimum on the potential energy surface – is not very useful. Flexible molecules change their conformation rapidly and are better thought of as having an ensemble of structures.
There are many tools for locating global minima and for sampling ensembles of molecules. These are not yet available as plug-ins in SEAMM, but hopefully will be soon.
Of course, the other limitation is the approximations inherent in DFTB+ and the parameterizations used. You should read about the science underpinning DFTB+, and the descriptions of the parameterizations at the DFTB website and references therein.