
Recipe 1: First Calculation with DFTB+ in SEAMM#
Tutorial#
This first recipe is a simple introduction to optimizing a structure – water – using DFTB+ and examining the output. In the recipe on the DFTB+ website you type in the coordinates for the atoms, use the mio-ext parameter set, and set up the optimization. We will do the same, but use the from-smiles-step to create the water molecules from its SMILES string:
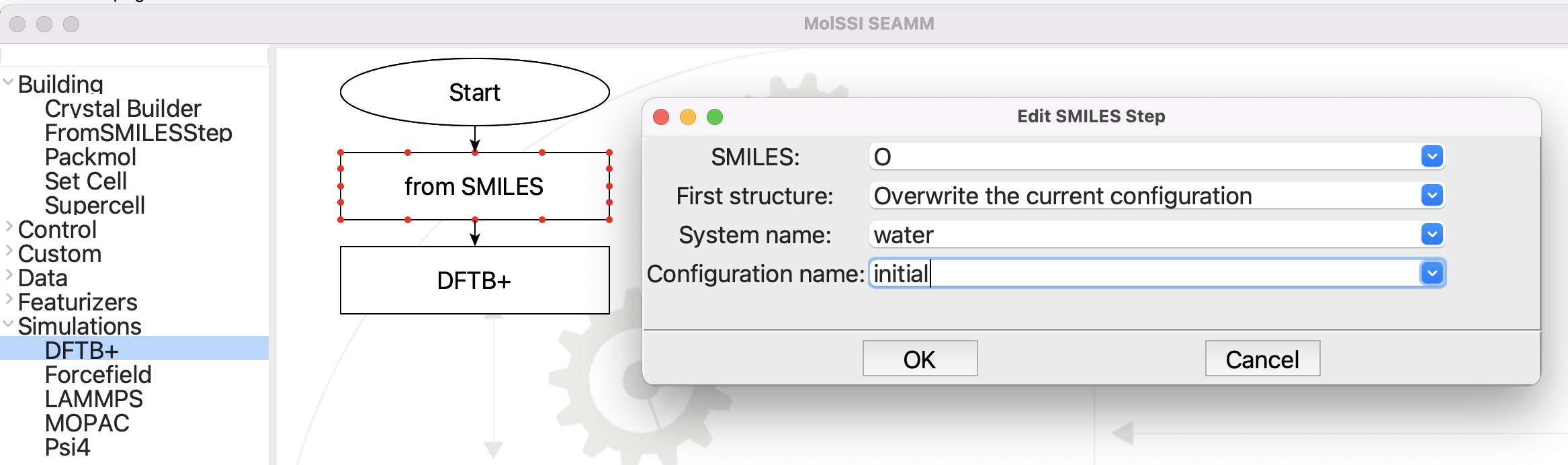
The flowchart and input for From SMILES#
The flowchart is very simple. It builds the structure from SMILES, and then runs DFTB+. The input for the From SMILES step is shown in the picture above. The only important parameter is SMILES, which is set to “O”, which is the SMILES representation for water.
Note
If you aren’t familiar with SEAMM and its flowcharts, there is a more gentle introduction in the SEAMM documentation.
If you are not familiar with SMILES, it is a convenient representation of moleculaes particularly organic molecules. It was created by Daylight Chemical Information Systems, which has a nice tutorial The EPA, which is where the original work on SMILES was done, also has nice introduction. Most databases of molecules have SMILES, as does Wikipedia, so using SMILES is a conveninet way to create molecules.
The DFTB+ flowchart is also very simple:
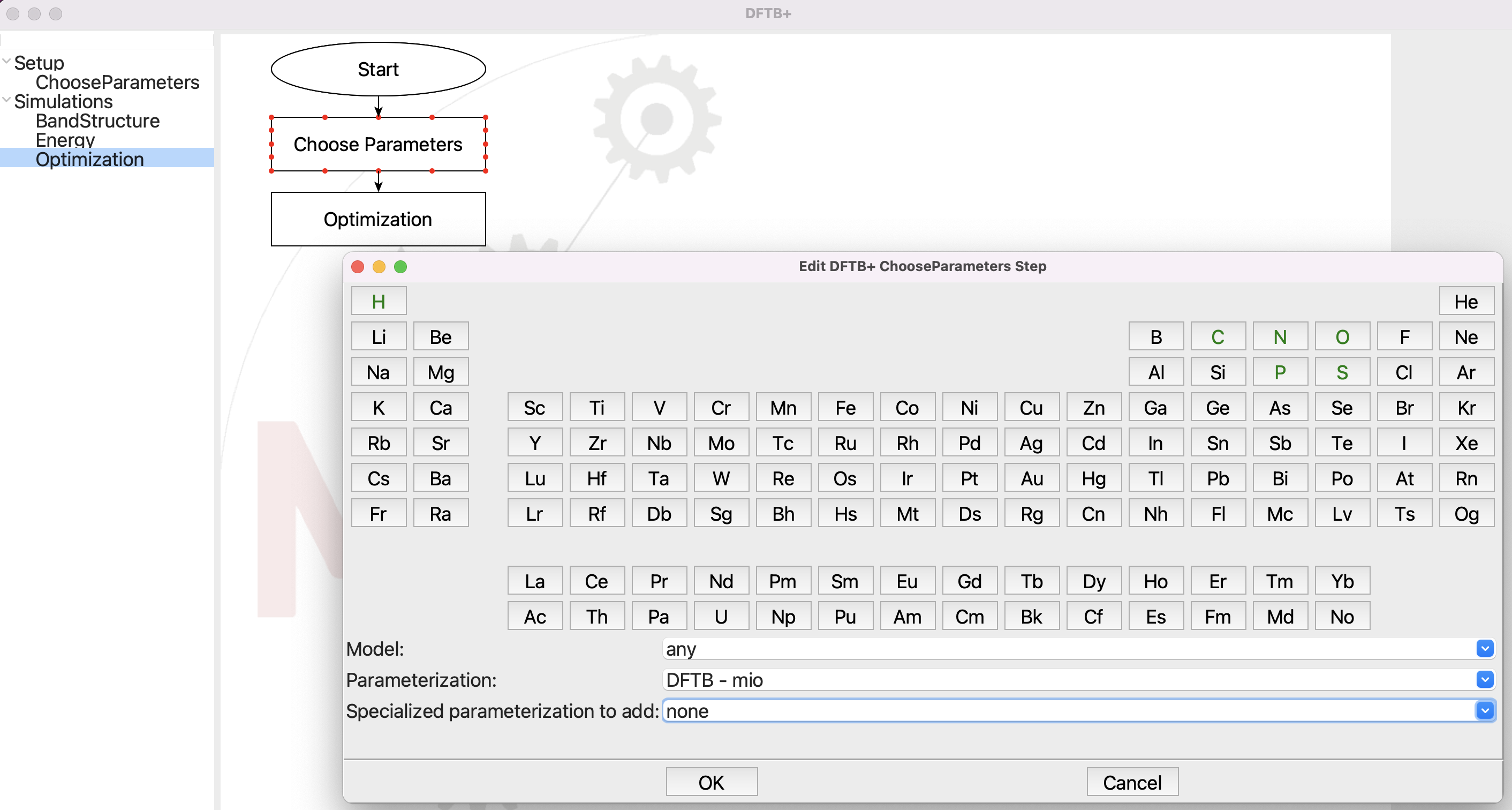
Choosing the parameters for DFTB+.#
The DFTB+ step in SEAMM uses the newer 3ob parameter set by default, so you need to change to the mio set as shown in the dialog.
The defaults for the optimization will work fine, so there is no need to make any changes:
The DFTB+ subflowchart is also very simple:
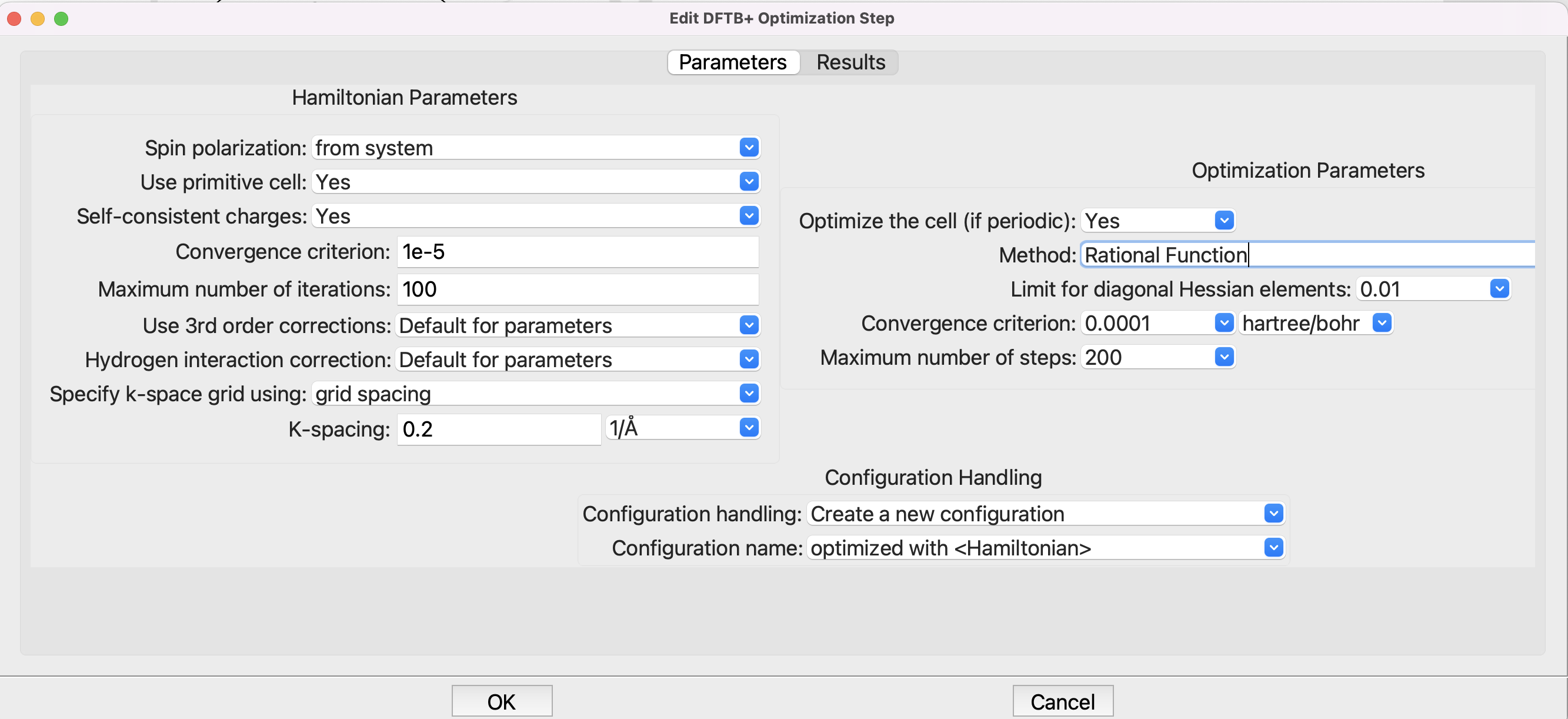
Optimization setup for DFTB+.#
Note that the original recipe uses the Conjugate Gradient driver, which is being phased out, so we will use the default Rational Function optimizer.
When you have finished the DFTB+ subflowchart, close it and then run the calculation using File / Run or ctrl-R (cmd-R on a Mac):
Running the job.#
Now head over to your browser and pull up the Dashboard. Find the job that you just ran, so that you can examine the input and output files. Here it is in my list of jobs in the Dashboard:

List of Jobs in DashBoard#
Click on the Job number in the left column (41 in the example) to go to the job’s page:
The main page for the job#
The left pane shows the list of files and folders in the Job. If you click on one of the files it will be displayed in the right pane. final_structure.mmcif shows the last structure:
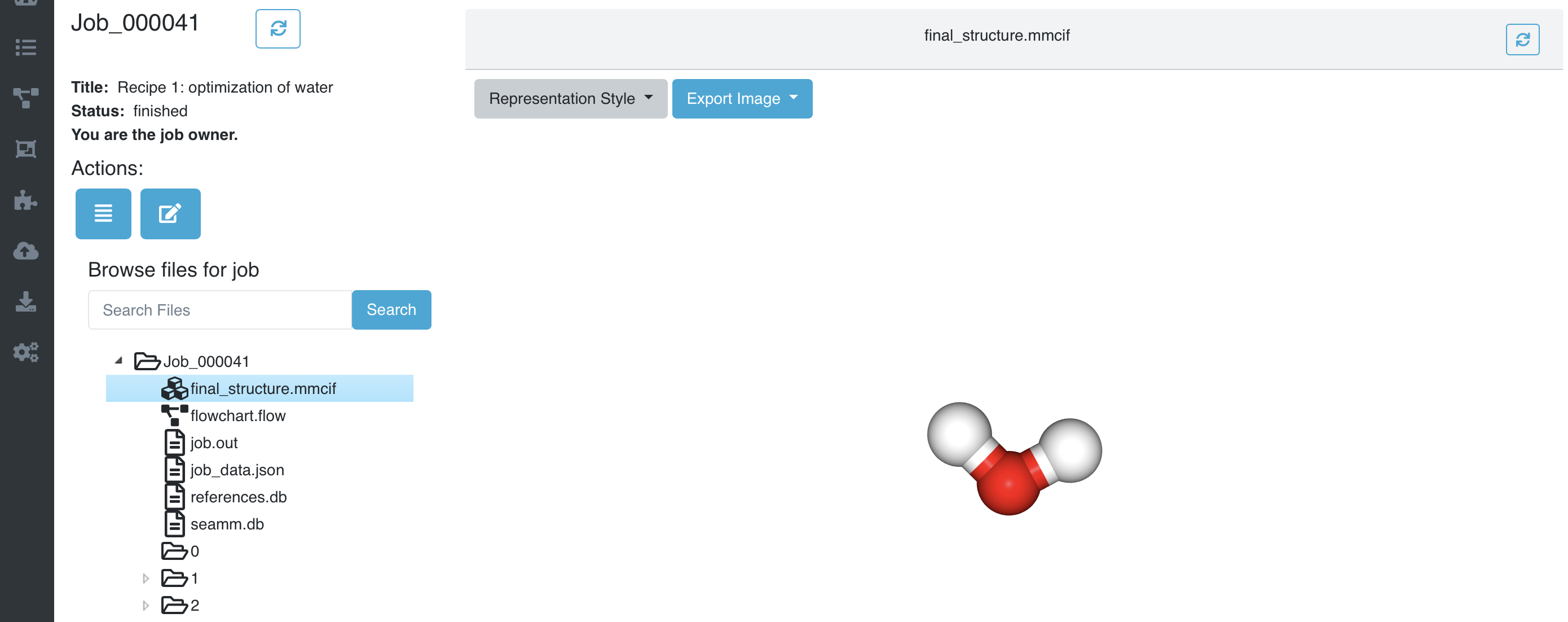
The optimized water molecule#
That looks right! You can rotate the picture by dragging the mouse over it.
Look at the job.out file next. This has the principle output of the job, plus any warnings or errors. The first part is a summary of the job that is written before any calculations are run so that you can see what it is going to do before it runs a long time – though our tiny job finished almost immediately:
Running in directory '/Users/psaxe/SEAMM/Jobs/projects/recipes/Job_000041'
Description of the flowchart
----------------------------
Step 0: Start 2022.7.25
Step 1: from SMILES 2021.10.13
Create the structure from the SMILES 'O', overwriting the current
configuration. The name of the system will be water. The name of the
configuration will be initial.
Step 2: DFTB+ 2022.7.24
Step 2.1: Choose Parameters
Using the 'DFTB - mio' set of Slater-Koster parameters.
Step 2.2: Optimization
Structural optimization using the Rational Function method with a
convergence criterion of 0.0 hartree/bohr. A maximum of 200 steps will be
used.
Doing a self-consistent charge calculation with a convergence criterion of
1e-05 charge units and a limit of 100 iterations. Whether to do a 3rd order
calculation and if so, what type, will be determined by the parameter set
used. Whether and how to correct interactions with hydrogen atoms will be
determined by the parameter set used. Any spins will be optimized. For
periodic system a Monkhorst-Pack grid with a spacing of 0.2 1/Å will be
used.
The next part shows what actually happened:
Running the flowchart
---------------------
Step 0: Start 2022.7.25
Step 1: from SMILES 2021.10.13
Create the structure from the SMILES 'O', overwriting the current
configuration. The name of the system will be water. The name of the
configuration will be initial.
Created a molecular structure with 3 atoms.
System name = water
Configuration name = initial
Step 2: DFTB+ 2022.7.24
Step 2.1: Choose Parameters
Using the 'DFTB - mio' set of Slater-Koster parameters.
Step 2.2: Optimization
Structural optimization using the Rational Function method with a
convergence criterion of 0.0001 E_h/a_0. A maximum of 200 steps will be
used.
Doing a self-consistent charge calculation with a convergence criterion of
1e-05 charge units and a limit of 100 iterations. Whether to do a 3rd order
calculation and if so, what type, will be determined by the parameter set
used. Whether and how to correct interactions with hydrogen atoms will be
determined by the parameter set used. Any spins will be optimized. For
periodic system a Monkhorst-Pack grid with a spacing of 0.2 1/Å will be
used.
The geometry optimization converged in 5 steps. The last change in
energy was 0.0 Eh.
The total energy is -4.077938 E_h. The charges converged to 0.000006.
The calculated formation energy is -284.2 kJ/mol.
Atomic charges
+--------+-----------+----------+
| Atom | Element | Charge |
|--------+-----------+----------|
| 1 | O | -0.59 |
| 2 | H | 0.3 |
| 3 | H | 0.3 |
+--------+-----------+----------+
Wrote the final structure to 'final_structure.mmcif' for viewing.
This is similar to the initial summary of the job, but this time it has detail about the number of atoms in the system that was built, how many iterations the calculation took, the energy -4.077938 E_h, etc.
If you read the DFTB+ tutorial, it shows the detailed output from DFTB+, and near the end is this section:
*** Geometry step: 12
iSCC Total electronic Diff electronic SCC error
1 -0.41505816E+01 0.00000000E+00 0.20115717E-02
2 -0.41505816E+01 -0.21681791E-07 0.14908557E-02
3 -0.41505816E+01 -0.26422777E-07 0.27122328E-07
Total Energy: -4.0779379339 H -110.9663 eV
Total Mermin free energy: -4.0779379339 H -110.9663 eV
Maximal force component: 0.280551E-05
>> Charges saved for restart in charges.bin
Geometry converged
Note that the energy given in job.out, -4.077938 E_h, is the same as the final energy in the original DFTB+ example, rounded to 6 decimal places. So we have reproduced the original recipe using SEAMM and get the same answer.
Note
If you want to see the input and output files for DFTB+, you can. They are in subfolders that correspond to the step number the flowchart. DFTB+ is the second step in the flowcharts, so open the folder ‘2’. The optimization was step ‘2.2’, so open the folder ‘2’ that is in the first folder ‘2’, i.e. 2/2.
job.out reports another energy, the formation energy -284.2 kJ/mol. This is almost the enthalpy (or heat) of formation, \(ΔH_f\), that chemists typically use. What is printed is actually \(ΔE_f\), i.e. the electronic energy of formation rather than the enthalpy, which includes other terms such as the zero-point energy and thermal energy. These terms tend to be relatively small, so \(ΔE_f ≈ ΔH_f\), and hence is quite useful.
Note
\(ΔE_f\) is calculated from the chemical reaction to create the compound from elements in their standard state. For water, the reaction is:
H₂(g) + ½O₂(g) --> H₂O(l)
which gives
The DFTB+ plug-in for SEAMM has tables of the energy of the elements in their standard state, calculated using the various parameter sets. It uses these energies in the equation above to calculate \(ΔE_f\). Not all element and parameter set combinations have been calculated yet, and occasionally there is a problem with the calculation, so some reference energies are missing, in which case the output let’s you know.
The final part of job.out is a list of references for the codes used, which are the appropriate citations for publications using the results:
Primary references:
(1) Jessica Nash and Eliseo Marin-Rimoldi and Mohammad Mostafanejad and Paul
Saxe. SEAMM: Simulation Environment for Atomistic and Molecular Modeling,
version 2022.7.25; The Molecular Sciences Software Institute (MolSSI):
Virginia Tech, Blacksburg, VA, USA, https://doi.org/10.5281/zenodo.5153984,
DOI: 10.5281/zenodo.5153984
(2) O'Boyle, Noel M. and Banck, Michael and James, Craig A. and Morley, Chris
and Vandermeersch, Tim and Hutchison, Geoffrey R. Open Babel: An open
chemical toolbox. Journal of Cheminformatics 2011, 3, 33. DOI:
10.1186/1758-2946-3-33
(3) Hourahine, B.; Aradi, B.; Blum, V.; Bonafé, F.; Buccheri, A.; Camacho, C.;
Cevallos, C.; Deshaye, M. Y.; Dumitrică, T.; Dominguez, A.; Ehlert, S.;
Elstner, M.; van der Heide, T.; Hermann, J.; Irle, S.; Kranz, J. J.; Köhler,
C.; Kowalczyk, T.; Kubař, T.; Lee, I. S.; Lutsker, V.; Maurer, R. J.; Min,
S. K.; Mitchell, I.; Negre, C.; Niehaus, T. A.; Niklasson, A. M. N.; Page,
A. J.; Pecchia, A.; Penazzi, G.; Persson, M. P.; Řezáč, J.; Sánchez, C. G.;
Sternberg, M.; Stöhr, M.; Stuckenberg, F.; Tkatchenko, A.; Yu, V. W.-z.;
Frauenheim, T. DFTB+, a software package for efficient approximate density
functional theory based atomistic simulations. The Journal of Chemical
Physics 2020, 152, 124101. DOI: 10.1063/1.5143190
Secondary references:
(1) Paul Saxe. From Smiles plug-in for SEAMM for creating structures from
SMILES, version 2021.10.13; The Molecular Sciences Software Institute
(MolSSI): Virginia Tech, Blacksburg, VA, USA, https://github.com/molssi-
seamm/from_smiles_step, DOI: 10.5281/zenodo.5159800
(2) Paul Saxe. DFTB+ plug-in for SEAMM, version 2022.7.24; The Molecular
Sciences Software Institute (MolSSI): Virginia Tech, Blacksburg, VA, USA,
https://github.com/molssi-seamm/dftbplus_step
This simple calculation doesn’t use many codes, but the list of references can become quite long. It is divided into sections based on a loose sense of importance to help you manage your citations.
Summary#
This job is as simple as it gets! Just two steps, one to build the structure and one to run the calculation, with all the parameters hard-wired. However, it already shows to power of SEAMM compared to running the calculation from the command-line. You didn’t need to edit the input file dftb_in.hsd and know the right keywords. Nor did you have to get the geometry of the molecule – though water is not that challenging.
You had to construct a flowchart, which was quite straightforward. You needed to enter the SMILES for water, but no geometry. Otherwise you could use the default parameters except for changing the parameter set from the default 3ob to the mio set to match the example from the DFTB+ website.
Mainly we focussed on looking through the output in the Dashboard. While you can see all the input to and output from DFTB+, we concentrated on the job.out file, which is a useful summary of the calculations and also contains other useful information.
In the next tutorial you will generalize this flowchart to make a more useful flowchart optimizing the structure of any (small) organic molecule.